
Abstract
Objective
Hurler Syndrome, (MPSIH) is an inborn error of glycosaminoglycan metabolism. Haematopoietic stem cell transplantation (HSCT) has transformed the prognosis for these children. Prior to transplant patients receive chemotherapy or chemo-radiotherapy. Regular screening for the development of endocrine sequelae is therefore essential. We present for the first time data on final adult height and endocrine complications in children with MPSIH post HSCT.
Design
Retrospective case note study and a prospective programme of growth and endocrine assessment.
Patients
22 patients were included, mean age at last assessment 12.2 (Range 6.3–21.6) years. Mean age at HSCT was 1.3 (SD 0.6) years. Conditioning included mostly busulphan and cyclophosphamide, with 5 patients receiving total body irradiation prior to second transplant.
Results
Height SDS decreased over time. Final height (FH) was attained in seven patients with male FH SDS −4.3 (Range −3.8, −5.1) and female FH SDS −3.4 (Range −2.9, −5.6). Eight of 13 patients tested had evidence of high growth hormone (GH) levels, while one had GH deficiency. Adrenal and thyroid function was normal in all. 11 patients were pubertal or post pubertal. Two females had pubertal failure requiring intervention. All male patients had spontaneous, complete puberty; however three patients have reduced testicular volumes. Five out of 13 patients tested had an abnormal oral glucose tolerance test.
Conclusion
Growth is impaired, primarily related to skeletal dysplasia, but also associated with GH resistance. Pubertal development may be compromised and abnormalities of glucose metabolism are common. We recommend a structured endocrine surveillance programme for these patients.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Hurler syndrome (MPSIH) is a lysosomal storage disorder caused by the accumulation of the glycosaminoglycans (GAGs), dermatan and heparan sulphate, due to deficiency of the lysosomal hydrolase α-L-iduronidase. The consequences of this deficiency are multisystem in nature and include skeletal deformities, corneal clouding, hepatosplenomegaly and profound psychomotor delay. Hurler syndrome was previously fatal by late childhood in the majority of cases. The outlook for these children has been transformed by the advent of haematopoietic stem cell transplant (HSCT) which is enabling children to live into adult life. Conditioning for HSCT involves chemotherapy for first transplants and historically chemo-radiotherapy, which may include total body irradiation (TBI) for repeat stem cell procedures where there is failure of the initial HSCT to engraft. As more children live longer, late effects of both the HSCT procedure and the natural history of the disease itself are becoming more apparent. Children with untreated mucopolysaccharidoses are known to develop build up of GAGS in selected endocrine glands (Schochet et al. 1974; Oda et al. 1988). In addition conditioning regimens for HSCT with or without the use of TBI as well as adverse effects such as graft versus host disease may all affect growth (Shalet et al. 1995; Ranke et al. 2005). Therefore one aspect of the long-term management of these patients should be growth and endocrine surveillance.
It is also recognised that HSCT does not alter the progression of the classical dystostosis multiplex, and therefore new challenges in the orthopaedic management of these patients have to be met. Our service has pioneered a multidisciplinary surveillance clinic to manage all long-term sequelae. Many of our patients are in late adolescence or early adulthood. As there are currently no published data on final height in MPS1H following HSCT, we have conducted an open study and describe the final adult height in members of this cohort, along with the other endocrine sequelae. We finally propose a scheme for screening and follow up in this complex patient group.
The study objectives were as follows:
-
1.
To evaluate growth patterns in terms of height, weight and body mass index.
-
2.
To describe the final height of patients who have completed longditudinal growth.
-
3.
To describe the endocrine co-morbidities encountered during surveillance of this cohort.
-
4.
To identify aetiological factors which may contribute to endocrine comorbidities in this group.
-
5.
To make recommendations for endocrine screening and follow up in patients with MPS1H.
Patients and methods
All surviving patients with MPSIH who had undergone HSCT were included in the study provided they were over 6 years of age at the time of last review. Height and weight data were taken from the patient’s clinic notes. Growth curves were created for each patient and from this height, weight and body mass index (BMI) at each year of age was calculated. Standard deviation scores (SDS) were calculated from the UK 1990 standards.
Subjects
A total of 22 patients (seven female) were eligible for inclusion in the study. Mean age at time of last assessment was 12.2 years (SD 4.4, range 6.2–21.6). Age at first HSCT was 1.3 years (SD 0.6, range 0.6–3.2). Nine patients required 2 HSCTs because of failure of engraftment or graft rejection, and of these, five patients had TBI as part of the conditioning for their second transplant. Conditioning regimens are shown in Table 1.
Surveillance assessments
Endocrine data were retrieved from case notes and laboratory reports. Sitting heights were not recorded, as the high prevalence of spinal deformities in this patient group makes this measurement very difficult to interpret. All parents of children had given consent for their child’s notes to be used for data acquisition.
The protocol for endocrine testing changed over the study period, with patients initially undergoing anterior pituitary stimulation tests pre- and post-transplant. No abnormalities were defined on pre-transplant testing. Therefore the subsequent protocol only included baseline post-transplant endocrine measurements. The current surveillance protocol is shown in Table 2.
Growth hormone deficiency (GHD) was defined as a peak growth hormone (GH) during an arginine or glucagon stimulation test of less than 15 mU/L (5.8 μg/L). High growth hormone levels were defined as a peak GH level >40 mU/L (15.4 μg/L) or a basal GH level >10 mU/L (3.8 μg/L) together with a peak GH >20 mU/l (7.6 μg/L) (Clayton et al. 2006). Hypothyroidism was defined as a baseline free (f)T4 below the normal range (<11 nmol/L) in the face of a normal TSH (0.1–5 mU/L) (secondary) or elevated TSH (>5 mU/L) (primary). Compensated hypothyroidism was defined as a TSH above the normal range with a normal fT4. Synacthen testing in the initial surveillance protocols was normal in all patients. Routine assessment of adrenal function was therefore discontinued, and only undertaken if deficiency was clinically suspected.
Delayed puberty was considered as onset of puberty after 13 years in girls (defined by attainment of breast stage 2) and 14 years in boys (defined by attainment of testicular volume of ≥4mls). Impairment of testicular / ovarian function was defined as LH or FSH above the normal range for the stage of puberty.
In view of the reported occurrence of abnormalities of glucose-insulin metabolism following TBI (Lorini et al. 1995; Taskinen et al. 2000; d'Annunzio et al. 2006; Shalitin et al. 2006; Chemaitilly et al. 2009), glucose tolerance tests were undertaken in the years post-transplant. Following an overnight fast, samples for glucose and insulin were taken before and 2 hours after ingestion of 1.75 g/kg glucose over 5 minutes (maximum 75 g glucose). Diabetes mellitus was defined as a fasting glucose level over 7 mmol/l or 2 hour glucose level over 11.1 mmol/l. Impaired glucose tolerance was defined as a 2 hour glucose level between 7.8 and 11.1 mmol/l. Impaired fasting glucose was defined as fasting glucose between 6.1–6.9 mmol/l with 2 hour glucose level less than 7.8 mmol/l (World-Health-Organisation 2006). Where appropriate, insulin resistance defined by HOMA IR was calculated using the computer model of Matthews et al. (Levy et al. 1998).
Results
Summary results for all patients are shown in Table 3.
Growth, final height and body mass index
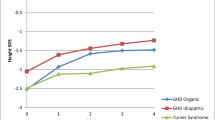
Final height was available in seven patients (three male, four female). Median (range) male final height was 137.5 cm (132,138.5), with median female final height being 143.1 cm (129, 145.8), giving a final height SDS of −4.3 in boys and −3.4 in girls. All patients had a final height < −2SD, and showed a gradual deceleration of all growth parameters with age as shown in Figs. 1 and 2. Mid parental heights (MPH) in this population were equivalent to the normal population (median [range] MPH SDS 0[−1.88, 2.37]). In regression analysis, number of transplants, use of TBI, genotype, degree of chimerism, donor source and post transplant enzyme levels did not influence height SDS. Body mass index declined over time from a median SDS of +2.2 two years post transplant to −0.04 ten years post transplant (see Fig. 2)

Growth curves for male and female patients with Hurler Syndrome post HSCT

Height, weight and BMI SDS for patients with Hurler Syndrome in the years following HSCT. Data are shown as median, interquartile range, 5th and 95th percentiles
Endocrine testing
Thirteen patients had GH stimulation tests post-transplant (Table 4). Eight of 13 had high levels of growth hormone, consistent with a degree of GH resistance. Of these, three patients were tested at more than one time point, two subsequently having normal GH levels and one having GH deficiency having been conditioned with chemotherapy only. When available, serum IGF 1 levels were below the mean for an age and sex matched reference range in all cases, with a value less than −2SD in 6/14. Three patients had high GH levels and low IGF1 providing further evidence consistent with biochemical GH resistance. Only one of these patients had received TBI.
All patients had their thyroid function checked. Three patients had a single mild elevation in their plasma TSH level which subsequently spontaneously normalised. No patient has required treatment for hypothyroidism.
Puberty
Of 22 children (seven female), 11 had progressed into or through puberty at ages within the normal range. Ten of the remaining children were prepubertal at appropriate ages, with one lost to follow up. Seven patients (mean age 18.5 years, four females) were post pubertal. Two of the girls had undergone a normal puberty, although one girl has a raised FSH (32 mU/L) in keeping with germinal epithelial damage from chemotherapy. The patient with normal gonadotrophins has subsequently conceived spontaneously (Hendriksz et al. 2004). The remaining two girls had pubertal arrest with elevated FSH levels (peak levels on gonadotrophin releasing hormone testing for patient: 2 LH 50.4 mU/L, FSH 77.7 mU/L, for patient 6: LH 4.7 mU/L, FSH 27 mU/L). Both required oestrogen supplementation to complete puberty, and one patient has a need for ongoing oestrogen replacement as a result of ovarian failure. This patient had received TBI for her second transplant. All three males completed a normal puberty, however two have small testes post-pubertally (maximum testicular volumes 10 ml) with currently normal basal and stimulated gonadotrophins levels for post pubertal males.
Of the four patients (mean age 13 years, all male) in the pubertal years, all underwent spontaneous puberty, however one patient has been found to have raised basal FSH on testing with small testes (testicular volumes 8 ml bilaterally at Tanner genital stage 4).
Glucose tolerance testing
Thirteen patients had been investigated with oral glucose tolerance tests (OGTT), of which eight had been tested on more than one occasion. Five patients (23% of the cohort) demonstrated an abnormality of glucose metabolism (Table 4). Two of these patients had received TBI of which one had progressed to type 2 diabetes mellitus and the other had evidence of insulin resistance with a HOMA IR of 3.4 at the age of 10 years (normal range <2 for age and sex (Almeida et al. 2008)) which has subsequently progressed to impaired glucose tolerance. All these abnormalities became apparent more than 8 years post transplant with the youngest patient being 10 years of age and 8.6 years post transplant. None of these 13 patients had chronic graft versus host disease (GVHD). Mean BMI SDS in these patients was +0.4, with only one patient with IGT having a BMI >+2. OGTT in the first 8 years following transplant was in four cases associated with low basal and stimulated insulin levels in the face of normoglycaemia (HOMA IR <0.5 in all cases). Subsequent testing after 8 years was normal in all with the exception of one patient who developed an impaired fasting glucose level; 86% of patients tested on more than one occasion demonstrated worsening of their glucose-insulin status over time
Discussion
Data on final height, pubertal progress and glucose-insulin metabolism in a population of children, adolescents and young people with MPSIH have been presented. Significant endocrine sequelae have been demonstrated; a tendency to develop insulin resistance, even in those who have received chemotherapy only regimens, marked short stature in the presence of normal/high GH secretion and below average IGF-I levels, impaired pubertal development in girls and reduced testicular volume in boys. We have not found any effects on the thyroid or adrenal axis.
As the success of HSCT as treatment for MPSH increases, so does the importance of monitoring for complications of this therapy. Children with this condition can look forward to a much improved prognosis, with the majority of transplant survivors capable of attending mainstream education often with additional help, as HSCT partially ameliorates the neurological deterioration seen without treatment. These children can now therefore be expected to live well into adult life. This study presents a large cohort of children with MPS1H, followed up over a longer period than previous studies, and is therefore able to look at the natural history of growth, puberty, endocrine and metabolic function. This study is limited by its retrospective nature, and protocols for testing have changed over the course of the study period. Nevertheless important data to inform future follow-up have been obtained.
There has been one previous study of growth and endocrine function in this population (Polgreen et al. 2008) who also reported evidence of short stature but were unable to comment on final adult height, instead noting that 87% of children had a height <−2SD by 10 years of age. This study did note that 27% had clinical or subclinical hypothyroidism - a finding not replicated in our cohort. Polgreen et al. also found that four patients (8%) of their whole cohort had a low peak GH and six patients (27%) had a low IGF-I level. A significantly greater proportion of patients received TBI (46%) in this study than in our population (22%) which is likely to account for these differences.
It is now clear that children with MPSIH post HSCT do have significant short stature, however stature is considerably greater than the 110 cm maximum height reported in a single child with Hurler syndrome without HSCT (Neufeld 2001). The terminal nature of MPS1H without transplant means that control data on final height are not available, however data of patients maintained on enzyme replacement therapy are available demonstrating a progressive reduction in height z-score over time (Tylki-Szymanska et al. 2010), although once again final height data are not presented. We have shown progressive growth failure through childhood, a lack of a pubertal growth spurt and a median final height of 137.5 cm (−4.34SD) in boys and 143.1 cm (−3.35SD) in girls. This gender difference is most likely due to artefact as a result of the small numbers involved. All patients who had completed their growth had a final height less than −2 SD, extending the findings of Vellodi et al. (which included some of the patients in this cohort) who found that mean height fell below the third centile by the age of five years, after an initial period of good growth (Vellodi et al. 1997). We have also found only minimal growth responses to recombinant human GH treatment in four patients, leading to discontinuation of treatment in three patients, in contrast to findings by Polgreen et al. who showed a modest initial increase in height velocity from 3.5 to 5.2 cm/year after one years treatment in eight children with MPS1H post HSCT (Polgreen et al. 2009). Our GH-IGF-I axis assessments pointed towards GH resistance, and these treatment trials indicate that this is not readily overcome by exogenous GH. It is recognised that TBI may be associated with GH resistance initially with evolution over time to GH deficiency. These patients potentially require on-going surveillance of GH status to detect late onset GHD in the early adult years, when treatment may be of benefit to bone accrual, body composition, muscle strength, lipid profiles and quality of life.
The reason for the ongoing short stature is likely to be multifactorial, however the greatest contribution to short stature in this group is likely to be the underlying skeletal dysplasia, dysostosis multiplex, which has been shown to progress despite HSCT (Field et al. 1994; Weisstein et al. 2004). The manifestations of this disorder include poor longitudinal growth in addition to genu valgum and spinal deformities which also contribute to short stature. Other factors which may well be related to the severity of dysostosis multiplex include genotype (Terlato and Cox 2003) and post treatment enzyme levels (Church et al. 2007) which in turn are affected by degree of chimerism and original donor source. Cord blood transplanted patients have been shown to have better engraftment and normal growth velocities in short term follow up (Staba et al. 2004). Although an initial period of good growth was also seen post HSCT, this was followed by a worsening of growth in later childhood (Vellodi et al. 1997). Regression analysis in our cohort did not identify any significant factors affecting final height possibly due to its small size, and the many possible aetiologies involved.
Transplant conditioning is also likely to play a part. It is well known that TBI can lead to short stature (Ranke et al. 2005), and chemotherapy only regimens have also been found to contribute to impairment of growth in otherwise healthy children post HSCT (Bakker et al. 2004). GVHD, particularly of the liver, by interfering with IGF-I production, could theoretically have contributed to the growth failure seen in our cohort, however significant chronic GVHD was only seen in one of our patients. What is not however in doubt is that patients undergoing HSCT for MPS1H have significantly shorter stature than those undergoing similar conditioning regimens for other indications even when TBI has been used, with patients following transplant for acute lymphoblastic leukaemia having only modest reductions in final height, and patients with thalassaemia showing improvements in height SDS post transplant (De Simone et al. 2001; Li et al. 2004; Bernard et al. 2009).
The pubertal development in our cohort is broadly similar to studies post HSCT in other conditions, which demonstrate a worse outcome in girls when compared with boys (Shalet et al. 1995; Couto-Silva et al. 2001). We found that all our patients entered puberty spontaneously although two females needed hormone supplementation to complete puberty, and had biochemical evidence of ovarian damage. This is in contrast to our male patients who had normal puberty both in timing and progression, although two patients had small testes postpubertally and one has small testes for his current pubertal stage. The young age at which children with MPSIH are transplanted is likely to be protective, since risk of gonadal damage increases with age, particularly in females. The finding of small testes in children undergoing HSCT has been previously noted (Haddy et al. 2009), particularly when TBI is used (Couto-Silva et al. 2001; Ishiguro et al. 2007). It has been noted that despite this, testosterone levels are sufficient to allow pubertal progression (Ranke et al. 2005) which is also what we have demonstrated. It has previously been noted that build up of GAGs can occur in the testes as well as other endocrine glands in untreated patients with mucopolysaccharidoses (Nagashima et al. 1976; Oda et al. 1988), however the early age of transplant in these patients (mean age of first transplant 1 year) would make this explanation less likely.
Our cohort demonstrated a high incidence of disturbances of glucose-insulin metabolism, with an overall incidence of disturbances of glucose metabolism of 23% despite an overall reduction in BMI SDS with time. There is increasing evidence for the development of features of the metabolic syndrome in patients who have received HSCT for other conditions, however this has not been described in MPS1H with or without transplantation. Shalitin et al. (2006) found a prevalence of 3.3% for type 2 diabetes in all patients undergoing BMT, and a similar number had impaired glucose tolerance. Likewise Lorini et al. (1995) demonstrated evidence of insulin resistance in survivors of HSCT as children, although their shorter follow up period (maximum follow up 10.2 years) may explain the lack of development of clinical diabetes or impaired glucose tolerance in this group. Shalitin et al. found an association with glucose intolerance and TBI, (in our cohort only two patients had received TBI). Taskinen et al. (2000) found an even higher prevalence of disturbances of glucose metabolism after HSCT of 52% over a follow-up period of 3–18 years in 23 patients treated in childhood for both malignant and non-malignant haematological conditions. Only four had received TBI in addition to chemotherapy. It will be important to continue surveillance for the development of glucose-insulin abnormalities in our MPS1H cohort and to look for this complication in cohorts of patients who have not been transplanted. In view of the poor growth, dysostosis multiplex with reduction in mobility, relative GH resistance, mild pubertal abnormalities and metabolic disturbances, we also recommend an assessment of bone density at the end of growth.
Conclusion
With children diagnosed with MPS1H now living into their adult years, we have shown that patients have significant short stature and considerably reduced final adult height. Patients suffer mild abnormalities of pubertal development, but there is a high incidence of abnormalities of glucose metabolism which has not been previously described. The cause of all these abnormalities is likely to be multifactorial and related to transplantation regimen in addition to genotype, post transplant enzyme levels and progression of dysostosis multiplex. Whilst advances in transplantation protocols have led to an increase in successful engraftment and reduction in risk, many of the endocrine morbidities in this patient group are still likely to be present. It is clear therefore that a structured screening programme for the detection and management of endocrine problems is required. In particular attention must be paid to the development of impaired glucose tolerance in this patient group from a young age. We would propose that screening (Table 2) using glucose tolerance testing is performed biannually from the age of 10 years, and that these patients are assessed twice yearly with growth and pubertal assessments. In view of the poor growth, dysostosis multiplex with reduction in mobility, relative GH resistance, mild pubertal abnormalities and metabolic disturbances, we also recommend an assessment of bone density at the end of growth. Together this schedule of monitoring and targeted screening will ensure that endocrine abnormalities can be rapidly identified and appropriate treatment given.
References
Almeida CA, Pinho AP, Ricco RG et al (2008) Determination of glycemia and insulinemia and the homeostasis model assessment (HOMA) in schoolchildren and adolescents with normal body mass index. J Pediatr Rio J 84(2):136–140
Bakker B, Oostdijk W, Bresters D et al (2004) Disturbances of growth and endocrine function after busulphan-based conditioning for haematopoietic stem cell transplantation during infancy and childhood. Bone Marrow Transplant 33(10):1049–1056
Bernard F, Bordigoni P, Simeoni MC et al (2009) Height growth during adolescence and final height after haematopoietic SCT for childhood acute leukaemia: the impact of a conditioning regimen with BU or TBI. Bone Marrow Transplant 43(8):637–642
Chemaitilly W, Boulad F, Oeffinger KC et al (2009) Disorders of glucose homeostasis in young adults treated with total body irradiation during childhood: a pilot study. Bone Marrow Transplant
Church H, Tylee K, Cooper A et al (2007) Biochemical monitoring after haemopoietic stem cell transplant for Hurler syndrome (MPSIH): implications for functional outcome after transplant in metabolic disease. Bone Marrow Transplant 39(4):207–210
Clayton PE, Ayoola O, Whatmore AJ (2006) Patient selection for IGF-I therapy. Horm Res 65(Suppl 1):28–34
Couto-Silva AC, Trivin C, Thibaud E et al (2001) Factors affecting gonadal function after bone marrow transplantation during childhood. Bone Marrow Transplant 28(1):67–75
d'Annunzio G, Bonetti F, Locatelli F et al (2006) Insulin resistance in children and adolescents after bone marrow transplantation for malignancies. Haematologica 91(12 Suppl):ELT12; author reply ELT13
De Simone M, Verrotti A, Iughetti L et al (2001) Final height of thalassemic patients who underwent bone marrow transplantation during childhood. Bone Marrow Transplant 28(2):201–205
Field RE, Buchanan JA, Copplemans MG et al (1994) Bone-marrow transplantation in Hurler's syndrome. Effect on skeletal development. J Bone Joint Surg Br 76(6):975–981
Haddy TB, Mosher RB, Reaman GH (2009) Late effects in long-term survivors after treatment for childhood acute leukemia. Clin Pediatr Phila 48(6):601–608
Hendriksz CJ, Moss GM, Wraith JE (2004) Pregnancy in a patient with mucopolysaccharidosis type IH homozygous for the W402X mutation. J Inherit Metab Dis 27(5):685–686
Ishiguro H, Yasuda Y, Tomita Y et al (2007) Gonadal shielding to irradiation is effective in protecting testicular growth and function in long-term survivors of bone marrow transplantation during childhood or adolescence. Bone Marrow Transplant 39(8):483–490
Levy JC, Matthews DR, Hermans MP (1998) Correct homeostasis model assessment (HOMA) evaluation uses the computer program. Diab Care 21(12):2191–2192
Li CK, Chik KW, Wong GW et al (2004) Growth and endocrine function following bone marrow transplantation for thalassemia major. Pediatr Hematol Oncol 21(5):411–419
Lorini R, Cortona L, Scaramuzza A et al (1995) Hyperinsulinemia in children and adolescents after bone marrow transplantation. Bone Marrow Transplant 15(6):873–877
Nagashima K, Endo H, Sakakibara K et al (1976) Morphological and biochemical studies of a case of mucopolysaccharidosis II (Hunter's syndrome). Acta Pathol Jpn 26(1):115–132
Neufeld EMI (2001) The metabolic and molecular basis of inherited disease.McGraw-Hill, New York
Oda H, Sasaki Y, Nakatani Y et al (1988) Hunter's syndrome. An ultrastructural study of an autopsy case. Acta Pathol Jpn 38(9):1175–1190
Polgreen LE, Tolar J, Plog M et al (2008) Growth and endocrine function in patients with Hurler syndrome after hematopoietic stem cell transplantation. Bone Marrow Transplant 41(12):1005–1011
Polgreen LE, Plog M, Schwender JD et al (2009) Short-term growth hormone treatment in children with Hurler syndrome after hematopoietic cell transplantation. Bone Marrow Transplant 44(5):279–285
Ranke MB, Schwarze CP, Dopfer R et al (2005) Late effects after stem cell transplantation (SCT) in children–growth and hormones. Bone Marrow Transplant 35(Suppl 1):S77–S81
Schochet SS Jr, McCormick WF, Halmi NS (1974) Pituitary gland in patients with Hurler syndrome. Light and electron microscopic study. Arch Pathol 97(2):96–99
Shalet SM, Didi M, Ogilvy-Stuart AL et al (1995) Growth and endocrine function after bone marrow transplantation. Clin Endocrinol Oxf 42(4):333–339
Shalitin S, Phillip M, Stein J et al (2006) Endocrine dysfunction and parameters of the metabolic syndrome after bone marrow transplantation during childhood and adolescence. Bone Marrow Transplant 37(12):1109–1117
Staba SL, Escolar ML, Poe M et al (2004) Cord-blood transplants from unrelated donors in patients with Hurler's syndrome. N Engl J Med 350(19):1960–1969
Taskinen M, Saarinen-Pihkala UM, Hovi L et al (2000) Impaired glucose tolerance and dyslipidaemia as late effects after bone-marrow transplantation in childhood. Lancet 356(9234):993–997
Terlato NJ, Cox GF (2003) Can mucopolysaccharidosis type I disease severity be predicted based on a patient's genotype? A comprehensive review of the literature. Genet Med 5(4):286–294
Tylki-Szymanska A, Rozdzynska A, Jurecka A et al (2010) Anthropometric data of 14 patients with mucopolysaccharidosis I: retrospective analysis and efficacy of recombinant human alpha-L-iduronidase (laronidase). Mol Genet Metab 99(1):10–17
Vellodi A, Young EP, Cooper A et al (1997) Bone marrow transplantation for mucopolysaccharidosis type I: experience of two British centres. Arch Dis Child 76(2):92–99
Weisstein JS, Delgado E, Steinbach LS et al (2004) Musculoskeletal manifestations of Hurler syndrome: long-term follow-up after bone marrow transplantation. J Pediatr Orthop 24(1):97–101
World-Health-Organisation (2006) Definition and diagnosis of diabetes mellitus and intermediate hyperglycaemia
Acknowledgements
The support of the Manchester Biomedical Research Centre is acknowledged.
Funding
No funding received.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by: Frits Wijburg
Competing interests: Nothing to declare
Rights and permissions
About this article
Cite this article
Gardner, C.J., Robinson, N., Meadows, T. et al. Growth, final height and endocrine sequelae in a UK population of patients with Hurler syndrome (MPS1H). J Inherit Metab Dis 34, 489–497 (2011). https://doi.org/10.1007/s10545-010-9262-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10545-010-9262-8