
Summary
Objective:
Alkaptonuria (AKU) is a rare inborn error of metabolism of aromatic amino acids and considered to be an autosomal recessive trait caused by mutations in the homogentisate 1,2-dioxygenase (HGD) gene. A dominant pattern of inheritance has been reported but was attributed to extended consanguinity in many cases. However, we have observed a non-consanguineous family segregating AKU in a dominant manner over three generations.
Results:
All affected individuals presented with typical features of AKU including darkening of the urine, ochronosis, arthropathy, and elevated urinary excretion of homogentisic acid. Sequence analysis of the HGD gene from genomic DNA of two affected individuals, uncle and niece, revealed a heterozygous missense mutation (M368V) in the uncle that was not present in his niece. Microsatellite genotyping demonstrated that both were heterozygous at the HGD locus and shared one haplotype. This haplotype did not contain a detectable HGD mutation. The haplotype was also found in a healthy son of the niece, making a dominant HGD mutation unlikely. Moreover, sequencing of cDNA from lymphoblastoid cells of the niece did not reveal an HGD mRNA with a potentially dominant-negative effect.
Conclusion:
Rare causes of the uncommon AKU inheritance in this family have to be considered, ranging from the coincidence of undetectable HGD mutations to a dominant mutation of a second, hitherto unknown AKU gene.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Alkaptonuria (AKU) was the first human disorder proposed to be a recessive trait (Garrod 1902). This paradigmatic inborn error of metabolism is characterized by the triad of ochronosis, which refers to the dark pigmentation of connective tissue, arthropathy, and homogentisic aciduria. Ochronosis is rarely noticeable externally before the third decade, when a grey to bluish-black pigmentation appears at the ear cartilage and at the site where the musculus rectus oculi inserts into the sclera. Ochronotic osteoarthritis may resemble rheumatoid arthritis clinically and affects nearly all patients in later years. In the fifth to sixth decades, AKU patients may develop kidney stones and calcification of the cardiac valve and coronary artery (La Du 1995; Phornphutkul et al 2002).
Homogentisic acid (HGA, 2,5-dihydroxyphenylacetate) is a catabolite of the aromatic amino acids phenylalanine and tyrosine. Alkali-induced oxidation of this ‘alcapton’ causes blackening of the urine, which represents a pathognomonic sign of AKU (La Du 1995). Accumulation of HGA has been attributed to complete deficiency of homogentisate 1,2-dioxygenase (HGD) (La Du et al 1958).
The HGD gene was first identified in the mould Aspergillus nidulans, in which the metabolic pathway is completely conserved (Fernández-Cañón and Peñalva 1995; Granadino et al 1997). In AKU patients, missense, frameshift, and intronic mutations of the HGD gene have been found. However, HGD mutations were not detectable in 9% of the AKU alleles (Beltran-Valero de Bernabe et al 1998).
Structural analysis of HGD revealed a hexamer of subunits arranged as a dimer of trimers. Many missense mutations are located in the region of contact between the subunits (Titus et al 2000). The aku mouse model resulted from an hgd frameshift mutation in the C-terminal region that comprises the active site of the enzyme (Kress et al 1999; Manning et al 1999). Heterozygous mice showed half-normal hepatic enzyme activity and did not excrete HGA, while homozygous mice had residual catalytic activity of 5.9% and excreted large amounts of HGA (Montagutelli etal 1994). Data on HGD activity in heterozygous humans have not been reported. Loading tests with tyrosine or HGA did not indicate a difference in metabolic activity between relatives of patients and normal controls. Thus, an oral load of 5 g HGA did not result in abnormal HGA excretion (reviewed in La Du 1995).
Apparently dominant inheritance in AKU has been reported before (Milch 1955). In some AKU families (Khachadurian and Abu Feisal 1958) this segregation pattern could be attributed to extended consanguinity (La Du 1995). Whether there are AKU cases with truly dominant mode of inheritance is still under debate. As yet, sufficient evidence has not been provided (La Du 1995). Here, we report a non-consanguineous pedigree segregating AKU in dominant pattern.
Materials and methods
Patients
Two affected individuals of this German family were available for clinical and genetic examination. The phenotypes of two deceased affected individuals were ascertained through review of medical records. All participating individuals gave their written informed consent prior to the study. Genomic sequence data in the index patient (III1) and her uncle (II3) were confirmed in DNA from independent blood samples. Lymphoblastoid cell lines were established by transformation with Epstein–Barr virus.
Analysis of homogentisic acid in urine
Urinary excretion of homogentisic acid was carried out by gas chromatography–mass spectrometry according to standard protocols.
Sequencing
Genomic DNA and total RNA were extracted from whole blood and lymphoblastoid cells, respectively, using standard methods. RT-PCR was carried out using the gene-specific primer RTas (5′-CCACA GAACACCAGCAAGTTTATTG-3′) in the 3′UTR (Superscript first-strand synthesis system, Invitrogen, Karlsruhe, Germany) followed by amplification of the entire translated cDNA in one fragment (cdna1s 5′-TT CCTGAAGTCACCACACATAG-3′ and cdna1as 5′-CAATACTACCAGAAAGCATGAGAT-3′). In addition, the entire coding cDNA was amplified in the three overlapping shorter fragments. Sequencing of the coding regions of the HGD gene was carried out by fluorescent dideoxy sequencing of PCR products generated from genomic DNA or cDNA, respectively, on an ABI 3130xl genetic analyser (Applied Biosystems, Darmstadt, Germany). For sequencing of all 14 HGD exons and flanking intronic regions, primers were selected according to the GenBank 6 database (accession numbers AF000573 and AF045167).
Genotyping
For haplotype analysis, markers within the HGD locus were chosen from the Ensembl and GDB databases, respectively, based on the human genome sequence (build 36.2). Alleles were amplified from genomic DNA using fluorescently labelled primers and size-fractionated on an ABI 3130xl. Allele sizes were determined using the GenScan software (Applied Biosystems).
Results
Clinical and biochemical findings
The family was of German origin and consanguineous relationships were denied. Patient II3 was 69 years of age at the time of examination. Blackening of the urine was present from early childhood. Biochemical analysis revealed massive urinary excretion of HGA (>1 mol/mol creatinine), which normally is not detectable. Excretion of other organic acids was normal. Progressive ochronotic arthropathy manifested in the third decade, followed by ochronosis of ears and eyes (Fig. 1). Arthropathy involved both large as well as small joints including hips, knees, shoulders, intervertebral joints, symphysis, interphalangeal and metacarpal joints and reached a severe level in the fifth decade. Echocardiography revealed apical and anterior hypokinesia. In addition, he was found to have high blood pressure as well as hypercholesterinaemia.

Ochronosis of ear lobe and sclera in patient II3
Patient II2, the brother of II3, had died at the age of 52 years from ochronotic valvular and coronary heart disease. He also had typical signs of AKU such as darkening of the urine, ochronosis of the ear lobes, and severe vertebral arthropathy. The same signs were found in individual I1, the mother of II2 and II3, who died at the age of 76 years.
In patient III1, daughter of II2, AKU was suspected during infancy when blackening of the urine was observed. The diagnosis was confirmed biochemically on the basis of massive urinary excretion of HGA (5.2 mol/mol creatinine). Excretion of other organic acids was normal. At the age of 32 years she had intermittent mild to moderate arthropathy of the intervertebral joints but no external signs of ochronosis. Her two sons (IV1, IV2) were healthy and urinary excretion of HGA was undetectable.
Molecular findings
Sequencing of the HGD gene from genomic DNA in patient II3 and his niece III1 revealed a heterozygous, previously described, missense mutation (M368V; Beltran-Valero de Bernabe et al 1998) in the uncle II3 but not in the niece III1. In addition, the uncle was heterozygous for a common non-synonymous polymorphism (H80Q, rs2255543), while his niece was homozygous at this position.
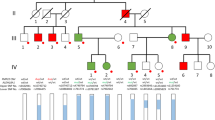
Haplotype analysis of the HGD locus was performed in affected and non-affected family members by genotyping of highly polymorphic microsatellite markers (Fig. 2). The constructed haplotypes revealed that all genotyped individuals were heterozygous at the HGD locus. Patient III1 shared one haplotype with her non-affected mother II1 and one haplotype with her affected uncle II3. Both haplotypes of her mother were different from those of her uncle. Thus, the microsatellite data did not support the possibility that identity-by-descent at the HGD locus (as caused by unrecognized consanguinity, for instance) had resulted in pseudodominance. Microsatellite data also excluded a large deletion of the HGD locus. In fact, the uncle II3 was heterozygous at three intragenic positions (D3S4497, M368V, rs2255543). Since the HGD enzyme functions as a complex multimer, we considered the possibility that a heterozygous intronic mutation, i.e., an aberrant splice product exerting a dominant-negative effect, could give rise to a defective enzyme variant. However, sequencing the entire HGD cDNA from lymphoblastoid cells of both affected individuals II3 and III1 did not reveal an HGD mRNA with a potentially dominant-negative effect. Moreover, the haplotype shared by II3 and III1 was also present in IV1 who had normal, that is, undetectable HGA excretion.

Pedigree with haplotypes at the HGD locus. The genotyped interval extended from 119.6 to 123.7 Mb on chromosome 3 with the HGD gene positioned at 121.8 to 121.9 Mb, and included two common intragenic markers, D3S4497/HGO-2 and rs2255543/H80Q, respectively. Patient II3 carried a heterozygous mutation M368V on haplotype 9-1-8-7-G-T-8-2-2 which was also found in his healthy daughter III3 but not in his affected niece III1. The other haplotype of II3 was present in III1 and in her healthy son IV1. Neither this haplotype nor the maternal haplotype of III1 included a detectable HGD mutation
M368V is considered to be the most prevalent HGD mutation in Europe (excluding the Slovak AKU patients) representing 20% of the AKU chromosomes (Beltran-Valero de Bernabe et al 1998; Zatkova et al 2000). With an estimated AKU incidence of 1:100 000–1:250 000, this implies an M368V allele frequency of 1:1600–1:2500. We sequenced exon 13 in 100 unrelated healthy individuals from Germany and detected M368V in 1 of the 200 chromosomes, suggesting a higher frequency of this variant in general or as a regional phenomenon (see Srsen et al (2002) for regional variance of AKU mutations).
Discussion
Several families have been reported before in which AKU appeared to be inherited dominantly (Milch 1955; Khachadurian and Abu Feisal 1958; La Du 1995). In some of them the seemingly dominant pattern of inheritance was attributable to extended consanguinity. However, haplotype data on the family presented here did not support the possibility that identity-by-descent at the HGD locus had resulted in pseudodominant alkaptonuria.
Sequencing the HGD gene in patient II3 (uncle of III1) revealed a heterozygous mutation (M368V) which has been implicated in AKU before (Beltran-Valero de Bernabe et al 1998). However, his also affected niece III1 did not carry this mutation. This indicates that M368V was not located on the HGD allele shared by uncle and niece, who both were heterozygous at the HGD locus as shown by haplotype analysis. Furthermore, M368V has not been reported to be dominant-negative. Thus, the heterozygous M368V mutation in the uncle cannot explain the occurrence of AKU in this family. Moreover, to our surprise, we found one heterozygous M368V carrier in 100 unrelated healthy controls. This finding may be accidental, but together with the fact that the recombinant M368V variant has 37% of the activity of the recombinant wild-type enzyme (Rodriguez et al 2000) it raises the question whether the effect of M368V is always pathogenic.
Apart from H80Q (rs2255543), a previously reported non-synonymous neutral variant (Beltran-Valero de Bernabe et al 1998), we did not find any further HGD mutation in this pedigree. Therefore, in order to uphold the hypothesis that the family’s AKU is caused by recessive HGD mutations, the coincidence of three rare events would have to be assumed, that is: (i) an undetectable mutation of the HGD allele that is shared by II3 and III1, (ii) an undetectable mutation of the allele that III1 received from her mother II1, and (iii) an additional HGD mutation in the non-consanguineous grandmother I1, who also had AKU.
It may also be possible that co-inheritance of two heterozygous mutations in different genes involved in aromatic amino acid metabolism, each causing a partial defect in the same pathway, could have resulted in the AKU phenotype. This phenomenon has been described for inherited enzyme deficiencies of mitochondrial fatty acid beta-oxidation and is referred to as synergistic heterozygosity (Schuler et al 2005). However, the absence of any detectable HGD mutation segregating with AKU in the pedigree presented here renders this possibility also unlikely.
Alternatively, a dominant HGD gene mutation may be hypothesized. Assume a dominant-negative allele that results in an HGD monomer that is incorporated randomly and inhibits all HGD hexamers in which it is contained. Then, only 1 of 26, that is, 1 of 64 hexamers would be active, corresponding to an enzyme activity of 1.6%, which is below the residual activity of 5.9% seen in aku mice (Montagutelli et al 1994). However, HGD deficiency is regarded as a strictly recessive disorder since none of the known HGD mutations has a dominant-negative effect, including missense mutations such as M368V, even if they are predicted to impair multimer assembly (Rodriguez et al 2000; Titus et al 2000). Our molecular data on cDNA from patient cells also did not produce any evidence of a potentially dominant-negative HGD gene product. Moreover, all three haplotypes found at the HGD locus of the two affected individuals, were present in healthy descendents (Fig. 2). A dominant HGD mutation therefore is unlikely in the present pedigree.
Thus, AKU in the family reported here may be caused by a dominant mutation of a different gene. We can only speculate on the altered function of the putative gene product. It is possible that the catalytic activity of HGD requires a hitherto unrecognized cofactor. Alternatively, this type of AKU may be due to an altered feedback control of HGA metabolism. In Pseudomonas putida HGA is a central intermediate of tyrosine catabolism and induces homogentisate dioxygenase (hmgA) (Arias-Barrau et al 2004). However, in the eukaryote mould Aspergillus nidulans, transcription of hmgA is induced by tyrosine and some of its metabolites but not by HGA itself (Fernández-Cañón and Peñalva 1995); and in fah −/− hpd −/− mice whose tyrosine metabolism is deficient upstream and downstream of HGD, administration of HGA even induces a twofold downregulation of the enzyme’s RNA (Tanaka et al 2006). Hence, there may be no simple HGA–HGD feedback control in eukaryote organisms. However, constitutional activation of any other negative control of HGD expression could possibly cause AKU. Screening of A. nidulans mutants, which led to the identification of HGD (Fernández-Cañón and Peñalva 1995), may also be helpful in identifying genes that regulate HGD expression.
Most disorders of metabolism have a recessive mode of inheritance. Kacser and Burns (1981) explained this mode by the distribution of ‘control’ on all enzymatic steps of a metabolic pathway, so that the fractional change in flux over the fractional change in activity is small for each individual enzyme. Typically, there is a hyperbolic relation between activity and flux with significant flux reduction occurring only close to zero enzyme activity. This is consistent with the finding in the aku mouse, in which a reduction of activity to 46% does not appear to change the flux while a reduction to 5.9% implies HGA accumulation (Montagutelli et al 1994). However, there are dominant metabolic disorders such as haploinsufficient disorders of lipoprotein metabolism and haem synthesis (Badminton and Elder 2005), or hawkinsinuria which results from a reactive intermediate that normally does not accumulate (Wilcken et al 1981). Other possible mechanisms of dominance have been proposed such as derangement of multimeric enzymes, disturbance of signalling pathways, and non-linear effects of haploinsufficient transcription factors (Veitia 2002; Wilkie 1994). One or other of these mechanisms might apply to the family reported here.
Abbreviations
- AKU:
-
alkaptonuria
- HGA:
-
homogentisic acid
- HGD:
-
homogentisate 1,2-dioxygenase
References
Arias-Barrau E, Olivera ER, Luengo JM, et al (2004) The homogentisate pathway: a central catabolic pathway involved in the degradation of l-phenylalanine, l-tyrosine, and 3-hydroxyphenylacetate in Pseudomonas putida. J Bacteriol 186: 5062–5077. doi:10.1128/JB.186.15.5062-5077.2004.
Badminton MN, Elder GH (2005) Molecular mechanisms of dominant expression in porphyria. J Inherit Metab Dis 28: 277–286. doi:10.1007/s10545-005-8050-3.
Beltran-Valero de Bernabe D, Granadino B, et al (1998) Mutation and polymorphism analysis of the human homogentisate 1,2-dioxygenase gene in alkaptonuria patients. Am J Hum Genet 62: 776–784. doi:10.1086/301805.
Fernández-Cañón JM, Peñalva MA (1995) Molecular characterization of a gene encoding a homogentisate dioxygenase from Aspergillus nidulans and identification of its human and plant homologues. J Biol Chem 270: 21199–21205. doi:10.1074/jbc.270.36.21199.
Garrod E (1902) The incidence of alkaptonuria: a study in chemical individuality. Lancet 160(4137): 1616–1620. doi:10.1016/S0140-6736(01)41972-6.
Granadino B, Beltrán-Valero de Bernabé D, Fernández-Cañón JM, Peñalva MA, Rodríguez de Córdoba S (1997) The human homogentisate 1,2-dioxygenase (HGO) gene. Genomics 43: 115–122. doi:10.1006/geno.1997.4805.
Kacser H, Burns JA (1981) The molecular basis of dominance. Genetics 97: 639–666.
Khachadurian A, Abu Feisal K (1958) Alkaptonuria; report of a family with seven cases appearing in four successive generations, with metabolic studies in one patient. J Chronic Dis 7: 455–465. doi:10.1016/0021-9681(58)90163-2.
Kress W, Schmidt SR, Halliger-Keller B, Montagutelli X, Müller CR (1999) The genetic defect of the alkaptonuric mouse (aku). Mamm Genome 10: 68–70. doi:10.1007/s003359900945.
La Du BN (1995) Alkaptonuria. In: Scriver CR, Beaudet AL, Sly W, Valle D, eds. The Metabolic and Molecular Bases of Inherited Disease, 7th edn. New York: McGraw-Hill, 1371–1386.
La Du BN, Zannoni VG, Laster L, Seegmiller JE (1958) The nature of the defect in tyrosine metabolism in alcaptonuria. J Biol Chem 230: 251–260.
Manning K, Fernandez-Canon JM, Montagutelli X, Grompe M (1999) Identification of the mutation in the alkaptonuria mouse model. Hum Mutat 13: 171. doi:10.1002/(SICI)1098-1004(1999)13:2<171::AID-HUMU15>3.0.CO;2-W.
Milch RA (1955) Direct inheritance of alcaptonuria. Metabolism 4: 513–518.
Montagutelli X, Lalouette A, Coude M, Kamoun P, Forest M, Guenet JL (1994) aku, a mutation of the mouse homologous to human alkaptonuria, maps to chromosome 16. Genomics 19: 9–11. doi:10.1006/geno.1994.1004.
Phornphutkul C, Introne WJ, Perry MB, et al (2002) Natural history of alkaptonuria. N Engl J Med 347: 2111–2121. doi:10.1056/NEJMoa021736.
Rodriguez JM, Timm DE, Titus GP, et al (2000) Structural and functional analysis of mutations in alkaptonuria. Hum Mol Genet 9: 2341–2350.
Schuler AM, Gower BA, Matern D, Rinaldo P, Vockley J, Wood PA (2005) Synergistic heterozygosity in mice with inherited enzyme deficiencies of mitochondrial fatty acid β-oxidation. Mol Genet Metab 85: 7–11.
Srsen S, Müller CR, Fregin A, Srsnova K (2002) Alkaptonuria in Slovakia: thirty-two years of research on phenotype and genotype. Mol Genet Metab 75: 353–759. doi:10.1016/S1096-7192(02)00002-1.
Tanaka Y, Nakamura K, Matsumoto S, et al (2006) Gene expression profiles of homogentisate-treated Fah−/− Hpd−/− mice using DNA microarrays. Mol Genet Metab 89: 203–209. doi:10.1016/j.ymgme.2005.09.022.
Titus GP, Mueller HA, Burgner J, Rodriguez de Cordoba S, Penalva MA, Timm DE (2000) Crystal structure of human homogentisate dioxygenase. Nat Struct Biol 7: 542–546. doi:10.1038/76756.
Veitia RA (2002) Exploring the etiology of haploinsufficiency. Bioessays 24: 175–184. doi:10.1002/bies.10023.
Wilcken B, Hammond JW, Howard N, Bohane T, Hocart C, Halpern B (1981) Hawkinsinuria: a dominantly inherited defect of tyrosine metabolism with severe effects in infancy. N Engl J Med 305: 865–868.
Wilkie AO (1994) The molecular basis of genetic dominance. J Med Genet 31: 89–98.
Zatkova A, Beltran Valero de Bernabe D, Polakova H, et al (2000) High frequency of alkaptonuria in Slovakia: evidence for the appearance of multiple mutations in HGO involving different mutational hot spots. Am J Hum Genet 67: 1333–1339.
Acknowledgement
We thank the family for participation in this study. We are grateful to Joachim Roesler, Department of Pediatrics, TU Dresden, for helpful discussions.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Communicating editor: Ertan Mayatepek
Competing interests: None declared
Rights and permissions
About this article
Cite this article
Oexle, K., Engel, K., Tinschert, S. et al. Three-generational alkaptonuria in a non-consanguineous family. J Inherit Metab Dis 31 (Suppl 2), 425–430 (2008). https://doi.org/10.1007/s10545-008-0994-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10545-008-0994-7