
Abstract
In order to study possible toxic side effects of potential drug compounds in vitro a reliable test system is needed. Predicting liver toxicity presents a major challenge of particular importance as liver cells grown in a cell culture suffer from a rapid loss of their liver specific functions. Therefore we are developing a new microfluidic test system for liver toxicity. This test system is based on an organ-like liver 3D co-culture of hepatocytes and endothelial cells. We devised a microfluidic chip featuring cell culture chambers with integrated electrodes for the assembly of liver sinusoids by dielectrophoresis. Fluid channels enable an organ-like perfusion with culture media and test compounds. Different chamber designs were studied and optimized with regard to dielectrophoretic force distribution, hydrodynamic flow profile, and cell trapping rate using numeric simulations. Based on simulation results a microchip was injection-moulded from COP. This chip allowed the assembly of viable hepatocytes and endothelial cells in a sinusoid-like fashion.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
It is important to have reliable high throughput toxicity test systems to develop drugs efficiently. Each year drug compounds need to be withdrawn from the market due to previously undiscovered side effects. This leads to a major risk for the patients’ health and the loss of large amounts of R&D investments. About 50% of all withdrawn drugs fail due to acute liver toxicity(Gershell and Atkins 2003). On account of this a new and better liver toxicity test system is required. Most in vitro tests used today rely on 2D-cell cultures which are insufficient to predict in vivo behaviour (Thomas et al. 2005; Gebhardt et al. 2003). Animal tests may lead to false results in consequence of species differences and should be avoided for ethical reasons. Ideally, a 3D-culture of human liver cells should be employed and would be expected to deliver reliable test results. There, liver cells would have to be arranged such as to mimic the organ morphology since hepatocytes require homogenous and heterogeneous cell-cell interactions as well as extra cellular matrix support to maintain their specific functionality. In addition, high throughput and automation of cell assembly, culturing and testing is imperative. Microfluidic system technology is particularly well suited to address these requirements.
Several microfluidic bioreactors and chips for in vitro liver culture have been developed over the last years. The group of L.G. Griffith used perfused multiwell plates with an integrated filter to accumulate rat liver cells and non-parenchymal cells (Powers et al. 2002; Sivaraman et al. 2005; Domansky et al. 2010). This bioreactor supported a viable culture for up to 7 days, allowed for high throughput and continuous perfusion of the culture. But still the in vivo liver 3D-structure, the sinusoid, which is important to support a stable hepatocyte phenotype (Poyck et al. 2008, Gebhardt and Hovhannisyan 2010), could not be maintained. Another approach to microfluidic hepatocyte cultures was recently shown by Chao et al. (Chao et al. 2009) where they used primary human hepatocytes. After first seeding cells onto a substrate and then assembling the microfluidic components the group cultured the cells for up to 24 h. Subsequently, they measured the hepatic clearance of several model drug compounds and compared the performance of this culture system to in vivo data. However, long term stability might prove a problem as the liver 3D-architecture is not preserved in this culture and endothelial cells were not included at all(Morin et al. 1986). Also the manual cell seeding and device assembly are laborious to perform. A microfluidic chip featuring automated seeding of hepatocytes into a 3D structure was developed by Toh et al. (Toh et al. 2009; Toh et al. 2007). They used a micro pillar array to capture freshly isolated rat hepatocytes and extra cellular matrix proteins. Their chip enables an organ-like perfusion and stable cell function was measured for 72 h. While the micro pillar structure resembles the in vivo liver structure to some degree, again the endothelial cells are missing. This mechanical trap also fails to separate living from dead cells. This separation, however, is important since cryopreserved human hepatocytes typically show an average viability of only about 50%. In contrast, by using dielectrophoresis as the driving force for cell manipulation the exclusive assembly of living cells can be achieved. Ho et al. (Ho et al. 2006) used a concentric electrode array to co-assemble cells from the human liver cell line HepG2 to human umbilical vein endothelial cells (HUVECs) in a liver lobule-like pattern. Their approach, however, does not allow for an organ-like perfusion. Also, dielectrophoresis with primary human cells was not demonstrated using this system.
The critical importance of mimicking the liver’s in vivo 3D-structure with its cell-cell and cell-matrix interactions has become evident in the recent years (Poyck et al. 2008; Gebhardt and Hovhannisyan 2010). To address this need we are developing a microfluidic chip which allows the assembly of human primary hepatocytes and endothelial cells into a liver sinusoid-like fashion (Fig. 1 (a)). It also combines the advantages of continuous perfusion, the selectivity dielectrophoresis for viable cells, the support by extra cellular matrix proteins, and automated cell seeding, culturing, and testing.

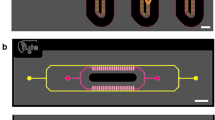
Concept of the HepaChip. (a) Principle of dielectrophoretic cell assembly in the chip. (b) Hepatocytes (orange) and endothelial cells (green) are arranged in a sinusoid-like fashion. The cell chamber features two electrodes on the outer channel walls for dielectrophoretic cell assembly, extra cellular matrix support in the cell seeding area and allows for an organ-like perfusion of the culture
2 Materials and methods
2.1 Principles of dielectrophoresis (DEP)
If particles such as cells suspended in a buffer solution are subjected to an inhomogeneous electric field, dipoles will be induced and a dielectrophoretic force arises which will act on the particles (Pohl 1978; Jones 1995; Pethig 1996; Pethig and Markx 1997) according to
Here, E is the amplitude of the ac electric field, R the particle radius, εm is the permittivity of the medium and Re[fCM(ω)] the real part of the Clausius-Mossotti factor. The Clausius-Mossotti fCM(ω) factor depends on the complex permittivity of the particle and the medium, respectively, as well as on the capacitance of the cell membrane (Fuhr et al. 1990). Re[fCM(ω)] defines the sign and thus the direction of the DEP force and depends on frequency of the electric field. If the particle is more polarisable than the medium, Re[fCM(ω)] is >0 and positive dielectrophoresis results. The force draws the particle into regions of large field strength. In case of negative dielectrophoresis the particle is less polarisable than the medium, Re[fCM(ω)] is <0 and the particle will be repelled towards regions of low field strength.
In insulator based dielectrophoresis (iDEP) insulator 3D-structures are used to form non-uniform electric fields (Masuda et al. 1989; Lee et al. 1994; Cummings and Singh 2003; Lapizco-Encinas et al. 2005; Lapizco-Encinas et al. 2004). As a particular advantage thereof cells will not get in contact with or be exposed to the electrode material.
2.2 Chip design and numeric simulations
For the assembly of liver-like cell structures a chip design based on iDEP was developed (Fig. 1 (a)). It comprises electrodes at the outer chamber walls and a variation of the channel cross-section in between. Thus, a field in homogeneity results giving rise to dielectrophoretic forces pointing towards the assembly gaps. Cells will be diverted into the assembly gaps as outlined in Fig. 1 (a). By assembling hepatocytes (orange cells in Fig. 1 (b)) and endothelial cells (green cells in Fig. 1 (b)), respectively, a liver sinusoid-like cell pattern is created. A coating of extra cellular matrix proteins on the gap surface will enable cell adhesion in this area. Micro pillars located at the front and back end of the assembly gaps represent flow barriers which provide for reduced flow velocity and a reduction of shear forces acting on cells settled on the assembly gaps.
Numerical multiphysics simulation using CFD ACE+ (ESI Group) was employed to optimize the cell chamber design. Numerical grids for different 2D and 3D cell chamber designs were constructed and flow, electric field distribution as well as hydrodynamic and dielectrophoretic forces were calculated.
2D designs were simulated with an applied voltage of 100 Vpp and a Clausius-Mossotti factor of one. In order to compare the dielectric force distribution in various designs, we used the quantity ln[FDEP/R3] which is independent of particle size.
Considering both hydrodynamic and dielectrophoretic forces, particle trajectories were calculated in the 3D chamber designs according to:
Thus, different designs could be compared with respect to their cell trapping rate. The trapping rate is defined as the ratio of the number of trajectories ending on the assembly gaps (corresponding to cells trapped by dielectrophoresis) vs. the total number of trajectories entering the cell chamber.
2.3 Chip fabrication and handling
Based on the simulated designs, microchips (Fig. 2) were injection-moulded from COP by microfluidic ChipShop. Gold electrodes were fabricated by sputtering titanium/gold layers through shadow masks. Protein binding sites were created on the surface of the assembly gaps by irradiation by low wavelength (180 nm) UV-light prior to bonding of the cover. After UV-activation the chip was bonded with a COP foil as cover.

Microfluidic chip for dielectrophoretic assembly of liver like 3D-cell structures. Fluid channels and electrodes contact the cell chambers where an electric field can be applied. Cells will be assembled into the assembly gaps by positive dielectrophoresis
For bubble free filling the chip was placed in a beaker with deionised water and vacuum was applied. Subsequently, pressure was restored so that water was sucked into the channels.
The chip was inserted into a chip fixture which was designed such as to allow for simultaneous fluidic and electric contacting of the device. The fixture was connected to a syringe pump/valve system for fluid control and a function generator/amplifier system used to apply ac electric voltages of up to 300 Vpp. The experimental setup is shown in Fig. 3.

Experimental setup: the chip is inserted into a chip fixture, which is mounted onto a fluorescence microscope. It allows for fluidic and electric connection of the chip to a syringe pump and a function generator. A LabView® application allows for automated control of the hardware components. Fluorescence images are recorded with a CCD-camera
Prior to cell assembly the chip was rinsed with a solution of Collagen Type IV hbc (10 μg/ml, BD Bioscience, Cat. Nr. 354245, Lot. Nr. 34774) and Pluronic® F-127 (1 mg/ml, Sigma-Aldrich, Germany, Cat. No. P-2443) in HEPES (10 mM, pH 5). In this preconditioning step the protein binds to the UV-irradiated gap surfaces to enhance the cell adhesion while Pluronic® F-127 molecules form a cell repulsive layer on non irradiated surface areas. The method for structured biofunctionalisation is described in detail elsewhere (J. Schütte 2010).
2.4 Cells and cell culture
Cryopreserved human hepatocytes and endothelial cells were obtained from Prof. Gebhardt’s research group at the University of Leipzig.
Cryopreserved cells were allowed to thaw on ice. The cell suspension was diluted by adding cold thaw medium (William’s Medium E, supplemented with 0.1 μM dexamethasone, 2 mM glutamine, 100 nM insulin, 100 U/ml penicillin, 100 μg/ml streptomycin, 10% fetal calf serum (v/v), 100 μM trolox, 15 U/ml DNase1) in portions of 0.5, 1, 2, and 3 times of the starting volume in 3 min intervals. Cells were centrifuged (5 min, hepatocytes at 50 g, endothelial cells at 250 g) and suspended in plating medium (thaw medium without DNAse1). Cell vitality was quantified by trypan blue. Cells were stained by adding calcein AM (green for hepatocytes, red orange for endothelial cells). Subsequently, the plating medium was removed by centrifugation and the cells were resuspended in low conductivity dielectrophoresis medium (10% sucrose, 0.66 mM sodium pyruvate, 0.49 mM CaCl2, 0.06 mM MgCl2•7H2O, 1 g/l glucose) to yield a concentration of 1 × 106 cells/ml.
2.5 Dielectrophoretic cell assembly
The suspension of hepatocytes was flushed into the chip at a flow rate of 20 to 100 μl/min. The voltage was adjusted such as to yield a field strength of about 30 kV/m at a frequency of 350 kHz. The cells were assembled onto the assembly gaps by dielectrophoretic forces. In order to remove cells that had not been assembled into the assembly gaps the chip was rinsed with dielectrophoresis medium at a rate of 50 μl/min. After rinsing the chip both voltage and flow were turned off for 10 min to allow for a stabilisation of hepatocyte adhesion on the gap surfaces. In the following step the endothelial cell suspension was flushed into the chip at a rate of 20 μl/min and the voltage was turned on again with the same amplitude. After completion of the assembly of endothelial cells the voltage was turned off and the co-culture was perfused with plating medium at a rate of 3 μl/min.
3 Results and discussion
3.1 Numeric simulation of different cell chamber designs
Cell assembly requires the cells to be diverted from the flow by dielectrophoresis. The cells will only be trapped if the dielectrophoretic force exceeds the hydrodynamic forces dragging them towards the chamber outlet. First we calculated the dielectrophoretic force distribution in 2D cross sections for different cell chamber designs. Figure 4 shows the cross section of two different cell chamber designs in comparison. The first design (Fig. 4 (a)) comprises a structured bottom and top half of both cell chambers. In the second design (Fig. 4 (b)) only the bottom chip part exhibits a 3D-microstructure whereas the cover consists of a flat lid. As expected in both designs the highest dielectrophoretic force (red areas) is observed at the edges of the gap region where the channel’s restriction is maximal. However, the two designs differ in their distribution of the dielectrophoretic force FDEP outside the gap region. In the first design the magnitude of FDEP decreases rapidly with increasing distance from the gap region. In the second design the regions of high FDEP reach further into the flow channels. These results show that a design featuring a flat cover—which in addition simplifies fabrication of the device—should allow for the trapping of cells moving in the channel even farther away from the gap region when compared to the symmetric design. To further increase FDEP in the flow channels the chamber was optimized in terms of gap height and the angle of inclination (Fig. 5). We found that by increasing gap height (Fig. 5 (a)) and decreasing inclination angle (Fig. 5 (b)) FDEP further out in the channel increases.

Dielectrophoretic force in a cross section of the cell chamber (a) with structured lid, (b) with flat cover. Gap height was 100 μm for both designs. Electrodes are indicated by yellow bars at the outer channel walls. (applied voltage U = 100 V, Clausius-Mossotti factor c fm (ω) = 1)

Comparison of dielectrophoretic forces in cell chamber cross sections with (a) different gap heights of 20, 60, and 100 μm, and (b) different inclination angles of 60°, 45° and 30°. Electrodes are indicated by yellow bars at the outer channel walls. (applied voltage U = 100 V, Clausius-Mossotti factor c fm (ω) = 1)
The next step was to optimize the chambers with respect to hydrodynamic forces. To mimic the in vivo liver sinusoidal structure dimension of approximately 2 × 2 × 30 hepatocytes with a cell diameter of about 50 μm each we used 3D simulations to calculate flow traces and velocity distributions in two chamber designs. Each design featured one assembly structure with a gap region of 100 μm height, 100 μm width and 1,500 μm length. Micro pillars were introduced up- and downstream of the assembly structure to create an area of reduced flow. Figure 6 shows the cell chambers with calculated flow traces and velocities of particles entering near the chamber bottom. In a design with an inclination angle of 45° and a flat channel bottom (Fig. 6 (a)) particles moving along the bottom of the chamber do not reach the sphere of influence of the dielectrophoretic force. By using an inclination angle of 33° and an inclined channel bottom (Fig. 6 (b)) particles passing along the bottom will be uplifted into the region of increased dielectrophoretic force and assembled into the gaps.

Comparison of flow velocities and trajectories (a) in a chamber with flat channel bottom and (b) with inclined channel bottom. In both chambers the assembly gap is isolated from the flow by two micro pillars. Electrodes are outlined as yellow surfaces. No dielectrophoretic forces were considered in these simulations
Three assembly structures per chamber were implemented in order to provide space for a sufficient number of cells to be assembled (Fig. 7 (a)). The calculated flow velocities (Fig. 7 (b)) clearly show the areas of reduced flow (blue areas) resulting from the micro pillars. The dielectrophoretic force distribution (Fig. 7 (c)) shows that cell assembly should take place on all three assembly structures as force maxima are observed at the edges of the ridge at the bottom of each assembly gap. Figure 7 (d) shows the dielectrophoretic force distribution in a plane level with the ridge at the bottom surface of the assembly gap and parallel to the chamber bottom. Local force maxima are not only located at the assembly structures but also at the micro pillars on either of their sides.

Numerical simulation of a representative 3D chamber design. (a) 3D model of assembly chamber. (b) Flow velocity distribution inside assembly chamber at z = 160 μm, 10 μm above assembly structures. Blue areas indicate areas of low flow velocity. (c) Distribution of ln[FDEP/R3] across assembly structures inside assembly chamber. Red areas indicate areas of high dielectrophoretic force. (d) Distribution of ln[FDEP/R3] inside assembly chamber at height z = 160 μm
Five different design variations were studied with respect to the cell trapping efficiency (Fig. 8). From the plots in Fig. 8 three different kinds of trajectories may be distinguished: i) there are traces passing through the cell chamber and leaving the chamber at the exit, ii) other traces end at the micro pillars’ heads and iii) the third kind of trajectories end on the ridge of the assembly structures. Applied to cell trapping, in case i) cells will not be assembled by dielectrophoresis but pass through the chamber because of the limited reach of the dielectrophoretic forces into the channel. In case ii) cells are trapped at the micro pillars, which is undesirable. However, since this is not an area of reduced flow these cells will be washed away after turning off the electric field. Yet, as these cells are lost, this will reduce the total number of cells that are actually available for assembly at the desired location inside the gaps, i.e. fraction iii).

Results of 3D multiphysics simulations of flow distribution and electric forces in various assembly chamber designs with particle trajectories calculated from hydrodynamic and dielectrophoretic forces
We compared five different micro pillar and assembly structure designs intended to reduce trapping at the micro pillars and increase the cell trapping rate on the assembly structures (Fig. 8). It turns out that 52 to 98% of the trajectories end inside the chamber depending on the type of design and the starting point of trajectories. Trajectories ending inside the chamber but not on the assembly structures are mostly ending in front of the micro pillars and at the electrode edges on both sides of the chamber. The overall cell trapping rates—i.e. fraction of cells being trapped on the assembly structures vs. total number of cells entering the assembly chamber—for the five designs are similar, ranging from 20% to 26,5% (Table 1). Thus, it is expected that the designs should not differ much in their cell trapping performance. For experimental verification chips featuring design 3, 4 and 5 were fabricated from COP to tested for cell assembly.
3.2 Comparison of simulation and experimental results of cell trapping
As predicted by simulations in all fabricated design cells could be withdrawn from the flow to trapped by dielectrophoresis. Figure 9 shows a typical result with an overlay of the simulated trajectories (green, design 5) and a fluorescence image of hepatocyte traces (white). It clearly shows the analogy between simulated and experimental particle trajectories under the influence of hydrodynamic and electric field forces.

Comparison of simulation and experimental results. Overlay of simulated particle trajectories (green traces) and a fluorescence image of the dielectrophoretic assembly of hepatocytes (white) in the chip. Location of electrodes is indicated by yellow areas
3.3 Dielectrophoretic assembly of liver sinusoid-like structures
We observed that only living cells (calcein positive) were addressed by DEP whereas dead cells moved through the chamber with the flow and were not affected by the electric field. In dead cells polarisability of the cytosol does not differ from the surrounding medium rendering the Clausius-Mossotti factor and the dielectrophoretic force zero. This is an important finding as cryopreserved hepatocytes show an average vitality of only about 50% after thawing. Therefore, the ability to actively select viable cells for cultivation is an important feature of this technology. Cell assembly could be shown for the HepG2 and HUVEC cell lines (data not shown) as well as for primary hepatocytes and endothelial cells. Figure 10 (a) shows the assembly of human primary hepatocytes. Upon application of the electric field the cells were drawn from the flow into the assembly gap. In less than 2 min the gap filled up with hepatocytes. Figure 10 (b) shows the assembly of primary human endothelial cells onto the adherent hepatocytes. The endothelial cells align to the hepatocytes forming so called pearl chains. After turning off the electric field and perfusing with culture medium the pearl chains dissolve and the cells rearrange into a more even distribution (Fig. 10 (c) and (d)) with the hepatocytes on the inside surrounded by endothelial cells. After turning off the electric field no cells were washed away by the applied flow. This shows that the adhesion to the collagen and the provision of an area of reduced flow by implementing the pillar structures are sufficient to support the cell culture.

(a) Assembly of primary human hepatocytes (green) into the assembly gaps (field strength: 30 kV/m, f = 350 kHz). (b) Assembly of primary human endothelial cells (red) upon adherent primary human hepatocytes (green) in the assembly gaps. (c) Assembled cells after turning off of the electric field. (d) Adherent cells after 10 min perfusion with plating medium. Direction of flow is indicated by arrows. In (c) and (d) the shape of the gap surface is outlined by dashed lines
4 Conclusion and outlook
By using multiphysics numeric simulations we were able to develop a microfluidic system which allows the assembly of primary human hepatocytes and endothelial cells in a liver sinusoid-like fashion by means of dielectrophoresis. We could show that only living cells are trapped thus only the healthiest cells will be used in culture. In particular, we were also able to co-assemble primary human hepatocytes and endothelial cells in liver sinusoid-like fashion and perfuse this co-culture afterwards.
The next step will be the demonstration of a long-term (>24 h) sinusoid cell culture on-chip. Liver metabolic functions (albumin synthesis, urea secretion, CYP activity) will be investigated. Potentially negative effects of the electric field and low conductivity medium on the cells need to be ruled out and optimal culture conditions (flow rate, media, and oxygen supply) will have to be identified. The HepaChip then will be evaluated and validated with respect to its efficiency in predicting liver toxicity and its application for substance screening.
5 Contributions of the authors
JS realised the dielectrophoretic cell assembly experiments. BH conducted the 3D simulations and programmed LabView software for operation of the fluidic controller. FH carried out the 2D simulations and contributed the proof of principle for the HepaChip together with MK. SW realised the fluidic and electric controller and designed the chip cassette. CF and KB contributed the cell culture and extra cellular matrix expertise. JB isolated the primary cells and developed the on chip hepatocyte media. RG contributed the liver expertise and participated in the development of the HepaChip concept. HB contributed to the chip design and participated in the development of the HepaChip concept. MS developed the concept and design of the HepaChip and directed the project at the NMI.
References
P. Chao, T. Maguire, E. Novik, K.C. Cheng, M.L. Yarmush, Biochem. Pharmacol. 78, 625–632 (2009)
E.B. Cummings, A.K. Singh, Anal. Chem. 75, 4724–4731 (2003)
K. Domansky, W. Inman, J. Serdy, A. Dash, M.H.M. Lim, L.G. Griffith, Lab Chip 10, 51–58 (2010)
G. Fuhr, P. Rösch, T. Müller, V. Dressler, H. Göring, Plant Cell Physiol. 31, 975–985 (1990)
R. Gebhardt, A. Hovhannisyan, Dev. Dyn. 239, 45–55 (2010)
R. Gebhardt, J.G. Hengstler, D. Muller, R. Glockner, P. Buenning, B. Laube, E. Schmelzer, M. Ullrich, D. Utesch, N. Hewitt, M. Ringel, B.R. Hilz, A. Bader, A. Langsch, T. Koose, H.J. Burger, J. Maas, F. Oesch, Drug Metab. Rev. 35, 145–213 (2003)
L.J. Gershell, J.H. Atkins, Nat. Rev. Drug Discov. 2, 321–327 (2003)
C.-T. Ho, R.-Z. Lin, W.-Y. Chang, H.-Y. Chang, C.-H. Liu, Lab Chip 6, 724–734 (2006)
T.B. Jones, Electro mechanics of particles (Cambridge University Press, Cambridge, 1995)
B.H. Lapizco-Encinas, B.A. Simmons, E.B. Cummings, Y. Fintschenko, Anal. Chem. 76, 1571–1579 (2004)
B.H. Lapizco-Encinas, R.V. Davalos, B.A. Simmons, E.B. Cummings, Y. Fintschenko, J. Microbiol, Meth. 62, 317–326 (2005)
S.W. Lee, S.D. Yang, Y.W. Kim, Y.K. Kim, in Eng. Med. Biol. Soc., Proc. 16th Annual International Conf. IEEE, Baltimore, MD, USA, 1994, pp. 1019–1020
S. Masuda, M. Washizu, T. Nanba, IEEE Trans. Ind. Appl. 25, 732–737 (1989)
O. Morin, C. Normand, J. Cell, Physiol. 129, 103–110 (1986)
R. Pethig, Crit. Rev. Biotechnol. 16(4), 331–348 (1996)
R. Pethig, G.H. Markx, Trends Biotechnol. 15, 426–431 (1997)
H.A. Pohl, Dielectrophoresis (Cambridge University Press, New York, 1978)
M.J. Powers, K. Domansky, M.R. Kaazempur-Mofrad, A. Kalezi, A. Capitano, A. Upadhyaya, P. Kurzawski, K.E. Wack, D.B. Stolz, R. Kamm, L.G. Griffith, Biotechnol. Bioeng. 78, 257–269 (2002)
P.P.C. Poyck, R. Hoekstra, J.L.M. Vermeulen, A. van Wijk, R. Chamuleau, T.B.M. Hakvoort, T.M. van Gulik, W.H. Lamers, Cells Tissues Organs 188, 259–269 (2008)
J. Schütte, C. Freudigmann, K. Benz, J. Böttger, R. Gebhardt, M. Stelzle, Lab Chip, 2010
A. Sivaraman, J.K. Leach, S. Townsend, T. Iida, B.J. Hogan, D.B. Stolz, R. Fry, L.D. Samson, S.R. Tannenbaum, L.G. Griffith, Curr. Drug Metab. 6, 569–591 (2005)
R.J. Thomas, R. Bhandari, D.A. Barrett, A.J. Bennett, J.R. Fry, D. Powe, B.J. Thomson, K.M. Shakesheff, Cells Tissues Organs 181, 67–79 (2005)
Y.C. Toh, C. Zhang, J. Zhang, Y.M. Khong, S. Chang, V.D. Samper, D. van Noort, D.W. Hutmacher, H. Yu, Lab Chip 7, 302–309 (2007)
Y.C. Toh, T.C. Lim, D. Tai, G. Xiao, D. van Noort, H. Yu, Lab Chip 9, 2026–2035 (2009)
Acknowledgements
Funding for this research was provided by the German Ministry for Education and Research, BMBF, through grant 01GG0729.
The authors thank Johanna Stelzle for proof reading and editing of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
Julia Schütte and Britta Hagmeyer contributed equally to this work
Rights and permissions
About this article
Cite this article
Schütte, J., Hagmeyer, B., Holzner, F. et al. “Artificial micro organs”—a microfluidic device for dielectrophoretic assembly of liver sinusoids. Biomed Microdevices 13, 493–501 (2011). https://doi.org/10.1007/s10544-011-9517-7
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10544-011-9517-7