
Abstract
New solvated Mo(VI) complexes were isolated from the reaction of [MoO2(acac)2] with asymmetric isatin bisthiocarbohydrazone ligands. The ligands were obtained from the reaction of isatin monothiocarbohydrazone with 3,5-dibromo salicylaldehyde (L1), 3,5-dichloro salicylaldehyde (L2) and 3-chloro-5-bromo salicylaldehyde (L3), respectively. In the complexes, the ligands serve as ONS donors and coordinate to the [MoO2]2+ nucleus. The bonding sites are azomethine nitrogen atom, phenolic oxygen atom and thiol sulfur atom. The sixth coordination site is completed by an oxygen atom from an ethanol solvent. The ethanol-coordinated Mo(VI) complexes, C1–C3, [MoO2L(EtOH)] (L: L1–L3), were characterized using elemental analysis, IR and 1H NMR spectroscopies, and conductivity measurements. By crystallizing ethanol-solvated solid complexes from an EtOH/DMSO mixture, DMSO-solvated complexes (C4–C6) suitable for X-ray crystallography were obtained. Crystal structure analysis supports the proposed complex structures and geometries, but the ethanol in the sixth coordination site has been replaced by DMSO. When the anticarcinogenic effects of the ligands and complexes (C1–C3) on the C6 cell line were examined, it was found that the complexes showed higher activity than the ligands. The C3 complex appears to have the best anti-cancer activity compared to doxorubicin. Additionally, all compounds were determined to have high total antioxidant capacity. Data obtained from theoretical studies (DFT and docking) support experimental studies.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Isatin (1H-indole-2,3-dione) is detected in humans as a metabolite of tryptophan or epinephrine and is widely distributed throughout the body within the central nervous system (Chowdhary et al. 2022). Isatin, found in biological systems, has many biological activities and also has very high anticancer potential. Generally, an increase in bioactivity is observed with complexation. Isatin derivatives target specific biomolecules or organelles as free ligands or metal complexes, showing promising antiproliferative properties against various cancer cells (Bugalia et al. 2023; Ferraz de Paiva et al. 2021). It has been determined that some isatin-β-thiocarbohydrazone compounds exhibit strong antitumor activity against cervical cancer (Hela) and kidney fibroblast cancer (COS-7) cell lines (Gabr et al. 2018).
Thiocarbohydrazones, are homologs of thiosemicarbazones, a class of compounds extensively studied for their antimicrobial, antifungal, and anticancer activities. Thiocarbohydrazone compounds with polydentate ligand properties have also been reported to possess a wide range of biological activities and applications, such as antiviral, antifungal, antibacterial, antimicrobial and anticancer activities (Abbas et al. 2013; Aldulaijan et al. 2023; Bonaccorso et al. 2020; Çavuş et al. 2024; Fouad et al. 2021; Hashim and Manoj 2023). In recent years, many asymmetric thiocarbohydrazone compounds have been synthesized and tested as cytotoxic and/or chemopreventive agents with strong antiproliferative activity against cancer cells (Gabr et al. 2017, 2018; Gangarapu et al. 2012; Hashim and Manoj 2023; Parsekar et al. 2022). They have also been evaluated for antimicrobial, antifungal, antiviral (against various DNA and RNA virus types) and antidiabetic activity (Abbas et al. 2013; Gabr et al. 2018; Gangarapu et al. 2014; Srividya and Reddy 2017; Tejasree et al. 2014; Yakan et al. 2022). Thiocarbohydrazone ligand systems have not been widely evaluated in the presence of metal ions or as isolated metal complexes. As metal-based drugs, it appears that there is untapped potential for this class of compounds (Bonaccorso et al. 2020). The attractiveness of thiocarbohydrazones for developing new metal-based drugs lies primarily in the presence of extra metal binding capacity (Bonaccorso et al. 2020). The presence of donor groups in the mononuclear complex formed as a result of complexation makes interaction with metal ions in biosystems possible.
Molybdenum is a vital trace element for humans, animals and plants and forms the active site of many enzymes (Odularu et al. 2019). Most enzymes and many potential catalysts that catalyze redox reactions contain a chelated [MoO2]2+ unit in their active site. ONS donor ligands constitute model systems for the active parts of molybdoenzymes such as xanthine oxidase and sulfite oxidase (Fayed et al. 2014; Hille 2013; Rana et al. 2002). Molybdenum atoms switch between IV and VI oxidation states during enzymatic reactions (Odularu et al. 2019). Additionally, molybdenum can mediate oxygen atom transfer (OAT) reactions at low potential (< 0 V) (Majumdar and Sarkar 2011). Coordination compounds of molybdenum are presented as potent anticancer and antimicrobial agents (Adam et al. 2022; Gretarsdóttir et al. 2016; Nair and Thankamani 2009; Saraswati and Kant 2013).
The aim of this study is to isolate potential bioactive Mo(VI) complexes from asymmetric bistiocarbohydrazone compounds where bioactive groups such as isatin and thiocarbohydrazide come together, to elucidate their structures and to examine their anticarcinogenic and antioxidant properties. Solid ethanol-solvated Mo(VI) complexes (C1–C3) of three asymmetric bisthiocarbohydrazone compounds prepared from the reactions of isatin monothiocarbohydrazone and 3,5-disubstituted salicylaldehydes were synthesized for the first time, and their structures were proposed using analytical and spectroscopic methods. By crystallizing the ethanol-solvated complexes (C1–C3) by slow evaporation in ethanol solution containing DMSO, DMSO solvated complex crystals (C4–C6) suitable for X-ray analysis were obtained. By analyzing their three-dimensional structures, the proposed complex structures and geometries were verified. In these complexes, ethanol in the sixth coordination was replaced by DMSO, which has higher donor properties. The antioxidant capacities and radical scavenging activities of the complexes were tested. The anticancer potentials all of the compounds were investigated against C6 cell line by XTT assay. Also, DFT and docking theoretical studies were performed.
Experimental
Materials and measurements
Thiocarbohydrazide and [MoO2(acac)2] were synthesized according to the described procedure in the literature (Burns 1968; Fernelius et al. 1960). Isatin, carbon disulfide, hydrazine hydrate, 3,5-dichloro salicylaldehyde, 3-bromo-5-chloro salicylaldehyde, 3,5-dibromo salicylaldehyde and solvents were used as received from Sigma Aldrich, Alfa Aesar and Merck chemical companies.
1H NMR spectra were recorded in DMSO-d6 on a Varian UNITY INOVA 500 MHz NMR spectrometer at room temperature. IR spectra were taken on an Agilent Cary 630 FTIR spectrometer in the 4000–650 cm−1 range. Elemental analysis was carried out using a Thermo Finnigan Flash EA 1112 Series Elemental Analyzer. UV–Vis spectra were measured in DMF using a Shimadzu 2600 UV–Vis Spectrophotometer. The magnetic measurements were carried out on a MK I model device obtained from Sherwood Scientific. The molar conductivities of the complexes in DMSO (10–3 M) were measured on a digital WPA CMD 750 conductivity meter at room temperature.
Synthesis of isatin monothiocarbohydrazone
Isatin monothiocarbohydrazone (L) was prepared according to a known method with minor modifications (Juranić et al. 1999). Spectroscopic data of the ligand are given in the Supplementary file.
Synthesis of asymmetric isatin bisthiocarbohydrazone ligands
The asymmetric isatin bisthiocarbohydrazone ligands (L1–L3) were synthesized from isatin monothiocarbohydrazone (L) using 3,5-dibromo salicylaldehyde, 3,5-dichloro salicylaldehyde, and 3-bromo-5-chloro salicylaldehyde, respectively, according to our previously published procedure (Kaya et al. 2021). Spectroscopic data of the ligands are given in the Supplementary file.
Synthesis of Mo(VI) complexes
Synthesis of C1, C2 and C3
The Mo(VI) complexes were prepared using the general procedure as given below.
1 mmol of ligand (L1-L3) was dissolved in 15 mL of ethanol. Then, 1 mmol of [MoO2(acac)2] was added to this solution and the mixture was heated at 60 °C for 3 h. The obtained orange product was filtered. The precipitate was washed with ethanol and dried in air.
[MoO2(L1)EtOH], (C1): Yield: 63%. Color: Orange. D. Temp.: > 300 °C. Anal. calcd. for C18H15Br2MoN5O5S (669.15 g/mol): C 32.31, H 2.26, N 10.47, S 4.79%. Found: C 32.03, H 2.19, N 10.17, S 4.63%. FT-IR (cm−1): ν(NH) 3172, ν(C=O)isatin 1692, ν(C=N) 1622 and 1588, ν(MoO2, asym. and sym.) 939 and 907. 1H NMR (500 MHz, DMSO-d6, ppm) δ: 13.26 (s, 1H, NH), 11.22 (s, 1H, isatin NH), 8.83 (s, 1H, CH=N), 8.00–6.93 (m, 6H, aromatic H), 4.30 (s, 1H, ethanol OH), 3.41 (q, 2H, ethanol –CH2–), 1.02 (t, 3H, ethanol –CH3). UV–Vis (DMF) [λmax (log Ɛ), nm (L mol−1 cm−1)]: 263 (4.41), 285 (4.25), 376 (4.52), 446 (4.40), 582 (3.88).
[MoO2(L2)EtOH], (C2): Yield: 56%. Color: Orange. D. Temp.: > 300 °C. Anal. calcd. for C18H15Cl2MoN5O5S (580.25 g/mol): C 37.26, H 2.61, N 12.07, S 5.53%. Found: C 37.11, H 2.24, N 11.98, S 5.27%. FT-IR (cm−1): ν(NH) 3213, ν(C=O)isatin 1693, ν(C=N) 1621 and 1581, ν(MoO2, asym. and sym.) 938 and 907. 1H NMR (500 MHz, DMSO-d6, ppm) δ: 13.26 (s, 1H, NH), 11.22 (s, 1H, isatin NH), 8.84 (s, 1H, CH=N), 7.81–6.93 (m, 6H, aromatic H), 4.30 (s, 1H, ethanol OH), 3.41 (q, 2H, ethanol –CH2–), 1.02 (t, 3H, ethanol –CH3). UV–Vis (DMF) [λmax (log Ɛ), nm (L mol−1 cm−1)]: 262 (4.34), 289 (4.13), 374 (4.49), 447 (4.35), 590 (3.22).
[MoO2(L3)EtOH], (C3): Yield: 65%. Color: Orange. D. Temp.: > 300 °C. Anal. calcd. for C18H15BrClMoN5O5S (624.70 g/mol): C 34.61, H 2.42, N 11.21, S 5.13%. Found: C 34.37, H 2.24, N 11.05, S 4.98%. FT-IR (cm−1): ν(NH) 3172, ν(C=O)isatin 1693, ν(C=N) 1623 and 1590, ν(MoO2, asym. and sym.) 940 and 909. 1H NMR (500 MHz, DMSO-d6, ppm) δ: 13.26 (s, 1H, NH), 11.22 (s, 1H, isatin NH), 8.84 (s, 1H, CH=N), 7.92–6.93 (m, 6H, aromatic H), 4.30 (s, 1H, ethanol OH), 3.41 (q, 2H, ethanol –CH2–), 1.02 (t, 3H, ethanol –CH3). UV–Vis (DMF) [λmax (log Ɛ), nm (L mol−1 cm−1)]: 262 (4.46), 286 (4.27), 375 (4.58), 440 (4.44), 588 (3.51).
Synthesis of C4, C5 and C6
[MoO2(L1)DMSO] (C4), [MoO2(L2)DMSO] (C5) and [MoO2(L3)DMSO] (C6) complexes were prepared in minimum yield as a result of the crystallization of ethanol solvate complexes (C1, C2 and C3) from EtOH/DMSO (10:1) solvent mixture by slow evaporation (after fifteen days).
X-ray data collection and structure refinement of C4, C5 and C6
Crystal data for C4, C5, and C6 were gathered with the help of a Bruker APEX-II CCD and MoK (λ = 0.71073 Å) radiation. Indexing of the data, integration, and absorption correction were all carried with the use of APEX, version 2014. Crystal structures were solved using SHELXT (Sheldrick 2015a) and then refined using the SHELXL (Sheldrick 2015b) in the Olex2 program package (Dolomanov et al. 2009). The asymmetric unit cell of C4 ([MoO2(L1)DMSO]) consists of two molecules, with the atoms C17, C18, S4 and C17A, C18A, S4A in DMSO molecule in one of them disordered over two sites with occupancies 0.85:0.15. In C5 ([MoO2(L2)DMSO]), the atoms S3/S4 in one coordinated DMSO molecule are disordered over two sites with occupancies 0.50:0.50. In C6 ([MoO2(L3)DMSO]), the atoms S1/S2 in one DMSO molecule disordered over two sites with occupancies 0.53:0.47. The disordered parts are shown in Figures S1–S3. The coordinates of aromatic CH and NH atoms were optimized geometrically using HFIX 43 code. The crystal data of C5 and C6 were non-merohedral twins, showing domains accounting for approximately 11 and 17%, respectively. The twin data of both structures were refined using a HKLF-5 file. Their displacement parameters were set to isotropic thermal displacement parameters (Uiso(H) = 1.2 × Ueq for CHaromatic and NH). Additional crystallographic data with CCDC reference numbers 2280901 (C4), 2280902 (C5) and 2280903 (C6) have been deposited within the Cambridge Crystallographic Data Center via www.ccdc.cam.ac.uk/deposit.
Computational details for density functional theory based analysis
DFT computations for studied ligands (L1, L2 and L3) and their Mo complexes (C1, C2 and C3) were performed with the help of Dmol3 module and Biovia Materials studio program. Optimizations of the studied chemical systems were made using the M06L functional and DND basis set (Rout et al. 2023). Chemical potential (µ), chemical hardness (η) and electronegativity (χ) are important parameters explaining the reactive or stable behavior of the chemical systems. Conceptual Density Functional Theory (CDFT) developed by Parr and coworkers (Chakraborty and Chattaraj 2021; Islam and Kaya 2018) includes simplified equations to compute the chemical reactivity descriptors and useful electronic structure rules. In this theory, chemical potential, chemical hardness and electronegativity are mathematically presented as:
There is an inverse relation between hardness and softness (σ) as given below:
In the given equations, E, N and ν(r) are total electronic energy, the total number of electrons and constant external potential, respectively. To relate aforegiven parameters to ionization energy (I) and electron affinity (A), a finite differences approach is applied to the parabolic relation between total electronic energy and the total number of electrons. To conclude, the following equations are derived for the computation of chemical potential, electronegativity and hardness (Berk et al. 2022; Kaya et al. 2016):
The electrophilicity index 1 (ω1) (Parr et al. 1999) and electrophilicity index 2 (ω2) (von Szentpály et al. 2020) are based on ground state parabola model and valence state parabola model, respectively. For the computation of these parameters, the following equations are used:
The prediction of electron donating and accepting abilities of the chemical species is very essential for the chemical reactivity analysis. Gázquez et al. (2007) introduced two new parameters named electrodonating power (ω−) and electroaccepting power (ω+) and proposed the following formula for the calculation of these parameters:
Net electrophilicity (\(\Delta \omega \pm\)) (Lebkiri et al. 2023) is calculated as:
In the theoretical part of this study, we considered Koopmans Theorem (1934) for the approximate prediction of ground state ionization energy and ground state electron affinity of the studied ligands and their Mo(VI) complexes.
Antioxidant capacity and activity tests
Different spectrophotometric methods were applied to investigate the antioxidant capacity and activities of the compounds. The CUPRAC method (Apak et al. 2004) was used to test each compound’s total antioxidant capacity. The basis of this procedure is the reduction of a copper(II)-neocuproine complex by antioxidants to a copper(I)-neocuproine complex. The procedure was carried out in accordance with our published study (Kaya et al. 2021). The obtained results are given in terms of “TEAC: Trolox Equivalent Antioxidant Capacity”.
The free radical scavenging activities of the compounds were measured according to the method of Brand-Williams et al. with minor modifications (Brand-Williams et al. 1995). The procedure was carried out in accordance with our published study (Kaya et al. 2021). The free radical scavenging activity was calculated according to the following equation:
% DPPH Radical Scavenging Activity = [ADPPH − AS/ADPPH] × 100, where ADPPH is the absorbance of the control solution and AS is the absorbance of the sample-radical mixture solution.
Cytotoxicity
The XTT test was used to determine cell viability (Roche Diagnostic, Germany). Before administration, L-759,633 was dissolved in dimethyl sulfoxide (DMSO) and diluted in DMEM. In 100 µl of DMEM culture media, cells were cultured in 96-well plates at a density of 1 × 104 cells per well and allowed to stick overnight. Doxorubicin was added to the C6 cell lines in increasing concentrations (100, 50, 25, 12.5, and 6.75 µM) and the plates were then incubated for 24, 48, and 72 h. The cells in the control groups were not given any treatment. After mixing, the absorbance of each well was measured at 450 nm and compared to the control using a microplate reader (Thermo, Germany). The cell viability was expressed as a percentage compared to the control group (100 percent of viability) using the following Eq. (1) (Joha et al. 2021):
Statistical analysis
The Student’s t-test or the ANOVA with multiple comparison post-hoc test were used to statistically evaluate the data. Data were shown as the standard deviations from the mean values. P value was less than 0.05 taken into account statistically. The experiments were conducted in triplicate.
Molecular docking
Molecular docking investigations of ligand–protein interactions were conducted using Molegro Virtual Docker (MVD) software (Bitencourt-Ferreira and de Azevedo 2019), which is known for its accuracy. The ligands were optimized using Density Functional Theory (DFT) using the M06L/DND basis set (via the use of COSMO as solvent—water) (Berisha 2019a, b; Klamt 2005; 2018) and then stored in the Protein Data Bank (PDB) format (Berman et al. 2003). The human c-Met in complex with the pyrido[2.3-d]pyrimidine analog III target was obtained from the protein data bank online site (http://www.rcsb.org/pdb/) using the PDB ID: 4GG7 (Wu et al. 2012). It was then produced on the MVD workspace by removing surplus water molecules and the cocrystallized ligand enclosed inside the crystal structure. The docking results were calculated using the MolDock score. The Discovery Studio v3.5 program was used to visualize different intermolecular interactions.
Results and discussion
Synthesis and some physical properties
In this study, asymmetric isatin bisthiocarbohydrazone ligands (L1–L3) were prepared by the reaction of 1-(2-oxoindolin-3-ylidene)thiocarbohydrazone (L) with respective 3,5-disubstituted salicylaldehydes in an ethanol-DMF mixture. New Mo(VI) complexes (C1–C3) were synthesized in ethanol using these asymmetric isatin bisthiocarbohydrazone ligands (L1–L3) and [MoO2(acac)2] (Scheme 1). The complexes are stable at room temperature and are soluble in acetone, DCM, DMSO and DMF. The elemental analyses are in good agreement with the molecular formula of the compounds. Low molar conductance values (11–19 Ω−1 cm2 mol−1) of the complexes in 10–3 M DMSO indicate the non-electrolytic nature of these complexes (Geary 1971; Kaya 2022). The μeff measurements suggest that the complexes contain molybdenum in the + 6 oxidation state with a d0 electronic configuration (El-Shahawi et al. 2019; Kaya et al. 2018).

Synthesis of the complexes (R1 = Br, R2 = Br for L1 and C1; R1 = Cl, R2 = Cl for L2 and C2; R1 = Br, R2 = Cl for L3 and C3)
Vibrational spectra
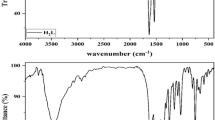
The bands around 3210 cm−1, attributed to the stretching vibration of OH in the ligands, disappear upon complexation, indicating the coordination of phenolate oxygen to the [MoO2]2+ core. The intensity of bands at 3176–3135 cm−1, belonging to the stretching vibration of two NH groups in the IR spectra of ligands, decreases after complexation. This shows that one of the NH groups in the thiocarbohydrazone molecule participates in the formation of the complex. Strong bands at around 1618 and 1600 cm−1 in the spectra of free thiocarbohydrazones are assigned to ν(C=N) stretches, one of which shifted to 1588, 1581 and 1590 cm−1, respectively, in the spectra of the complexes. This result indicates the coordination of azomethine nitrogen to the metal ion. The ν(C=O) stretch of the isatin fragment in the spectrum of the ligands appears almost in the same place in the spectra of the complexes, supporting that the isatin fragment does not participate in the formation of the complexation. Finally, the complexes exhibit two characteristic bands in the ranges 907–909 and 938–940 cm−1 for symmetric and asymmetric stretching vibrations of [MoO2]2+ core (Asha and Prathapachandra Kurup 2020; Kaya et al. 2021; Muğlu et al. 2021; Sathisha et al. 2008). IR spectra of the ligands and complexes are given in Figures S4–S5 and S11–S13.
1H NMR spectra
Thiocarbohydrazone has two tautomer structures: thion (C=S) and thiol (C–SH). The thione form (C=S) from these is in equilibrium. For the thiol tautomer form, which contains the C–SH structure, two isomeric configurations can be proposed. One of these is the Z (syn) form, where the central C=N bonds are in the same plane, and the other is the E (anti) form, where the C=N bonds are vertical to each other (Dragancea et al. 2005). Accordingly, in the NMR spectra of the ligands, NH signals of thiocarbohydrazide moiety were observed around 12.68, 12.51 ppm (s, isomer ratio: 1/1, 1H) and 13.02, 10.14 ppm (s, isomer ratio: 1/1, 1H). Similarly, OH and NH (isatin) signals were observed as two separate singlets. This isomerism disappears in the complexes due to the hindrance of rotation of the thiocarbohydrazone group around its double bonds. The fact that the signals observed for OH protons in the ligands do not appear in the spectra of the complexes supports the binding of the ligand to the metal ion via deprotonation of the OH group. The NH signal of isatin was observed as a singlet at almost the same place in the spectra of the complexes (11.22 ppm). A single NH signal of thiocarbohydrazide around 13.26 ppm was observed in the spectra of the complexes. The disappearance of the other NH signal of thiocarbohydrazide in the spectra of the complexes shows that one of the NH groups is bound to the metal ion by deprotonation. The absence of the other NH signal of thiocarbohydrazide in the spectra of the complexes shows that one of the NH groups is bound to the metal ion by deprotonation. These results support that half of the ligand is involved in the formation of the complex. In addition, signals belonging to ethanol in the spectra of the complexes were observed at 4.30 ppm (s, 1H, ethanol –OH), 3.41 ppm (q, 2H, ethanol –CH2–) and 1.02 ppm (t, 3H, ethanol –CH3), respectively (Gangarapu et al. 2014; Kaya 2022; Kaya et al. 2015, 2021; Muğlu et al. 2023). 1H NMR spectra of the ligands and complexes are given in Figures S7–S9 and S14–S16.
Electronic spectra
Electronic spectra of the ligands and complexes were measured in DMF at room temperature using 3 × 10–5 M solution. In the spectra of the ligands, the bands observed around 265 and 286 nm are attributed to π → π* transitions belonging to the phenyl rings. These bands were observed at almost the same location in the spectra of complexes. In the spectra of ligands, the bands at 375 and 453–458 nm are probably due to the n → π* transitions of the C=N and C=S groups. These bands were recorded at 374–376 nm and 440–447 nm in the spectra of complexes. This supports the coordination of nitrogen and sulfur atoms to the metal. The weak shoulder bands observed at 582–590 nm in the spectra of the complexes can be attributed to LMCT transitions (Ceylan et al. 2015; Kaya et al. 2021). UV–Vis spectra of the ligands and complexes are given in Figures S10 and S17.
Crystallographic descriptions
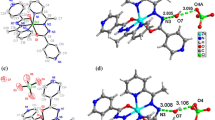
In the crystallization studies of ethanol-solvated complexes (C1, C2 and C3) in ethanol solvent, crystals suitable for X-ray structure analysis could not be obtained. Crystallization of these solid complexes from an EtOH/DMSO (10:1) mixture resulted in the formation of DMSO-solvated complexes (C4, C5 and C6), and their structures were determined by X-ray diffraction method. The information on the refining of all crystallographic data is displayed in Table 1. Table S1 contains all of the important bond distances and angles. All of the Mo(VI) complexes crystallize in the triclinic space group P-1, and their asymmetric unit is composed of two crystallographically distinct molecular complexes (Figs. 1A, 2A and 3A). In each of these complexes, the Mo(VI) atom adopts a distorted octahedral coordination geometry with the NO4S coordination sphere. The octahedral coordination geometry is completed by the DMSO molecule. Each dianionic tridentate thiocarbohydrazone-based ligand in C4, C5 and C6 is bonded to the Mo(VI) metal center through the phenolate oxygen, thiolate sulfur and the imine nitrogen. This results in the formation of a five-membered and a six-membered chelate ring with average S–Mo–N/O–Mo–N bite angles of 76.25(3)°–83.50(3)°, 76.20(3)°–82.60(4)°, and 76.11(3)°–82.55(3)° respectively. These values are almost identical to those seen in other Mo(VI) complexes in the literature (Rakshit et al. 2016; Rana et al. 2002; Roy et al. 2018). The values of the double M–O bonds in [MoO2]2+ in C4, C5, and C6 are in the range of 1.669(8)–1.703(8) Å, which are very common for Mo(VI) complexes (Eğlence-Bakır et al. 2019, 2021). Each ligand has a somewhat planar molecular configuration with quite small dihedral angles, in which the 2-oxoindolin-3-ylidene subunit and the phenyl ring (5.15°–9.70° for C4, 4.43°–7.38° for C5, 5.06°–8.31° for C6). In C4, weak intermolecular contacts such as C–H···Br (C2–H2···Br2, d(H···Br) = 2.859 Å, C13–H13···Br4, d(H···Br) = 2.807 Å) contribute to the formation of a 1D zigzag chain structure, in which very short distance π···π interactions with d(π···π) = 3.708(8) Å occur between the ligands. Additionally, a 3D supramolecular network of C4 is formed by numerous classical and non-classical hydrogen bonding interactions (N–H···O, C–H···O, C–H···S) such as N8–H8···O8, N5–H5···O5, N10–H10···O10, C35–H35B···O6, C35–H35B···O9, C36–H36B···O9, C6–H6···O4, C15–H15···O1, C17–H17C···S1, C35–H35A···S1, C22–H22···S4, C18–H18B···S3. Some hydrogen bonding and π···π interactions in C4 are shown in Fig. 1B. Abundant hydrogen bonding interactions (C25–H25···Cl2, C15–H15···Cl1, C13–H13···O4, C17–H17B···O7, C18–H18B···O7, N5–H5A···O9, N10–H10···O10, C17–H17B···S3, C20–H20B···S1, C19–H19A···S6, C17–H17C···S5 and C20–H20B···S1) play an important role in the crystal packing of C5. Also, a strong π···π interaction with d = 3.721(8) Å exists in C5. Some of these interactions are shown in Fig. 2B. In C6, classical N–H···O (N9–H9···O10, N10–H10···O9) and non-classical C–H···O (C19–H19···O5, C36–H36B···O2, C35–H35B···O2, C29–H29···O7), C–H···S (C35–H35A···S6, C24–H24E···S6, C23–H23E···S3, C5–H5···S1) and C–H···X (X: Cl, Br) interactions (C17–H17···Br2, C31–H31···Cl1) help create a 3D supramolecular network. Similar to C4 and C5, the impressive π···π interaction with d(π···π) = 3.761(7) Å is observed in the crystal structure of C6. Some of these interactions in C6 are illustrated in the Fig. 3B.

A ORTEP style drawings (50% probability level) of the molecular structures showing the corresponding the inter-ring dihedral angles between the planes in C4. B The intermolecular C–H···Br/O, N–H···O hydrogen bonding and the intermolecular π···π stacking interactions in C4 (a: C2–H2···Br2, d(H···Br) = 2.859 Å, b: C13–H13···Br4, d(H···Br) = 2.807 Å, c: N8–H8···O8, d(H···O) = 2.710 Å, d: C35–H35B···O6, d(H···O) = 2.506 Å, e: C35–H35B···O9, d(H···O) = 2.609 Å), f: C36–H36B···O9, d(H···O) = 2.625 Å), π···π, d(π···π) = 3.708(8) Å)

A ORTEP style drawings (50% probability level) of the molecular structures showing the corresponding the inter-ring dihedral angles between the planes in C5. B The intermolecular C–H···Cl/O/S hydrogen bonding and the intermolecular π···π stacking interactions in C5 (a: C25–H25···Cl2, d(H···Cl) = 2.895 Å, b: C15–H15···Cl1, d(H···Cl) = 2.853 Å, c: C17–H17B···O7, d(H···O) = 2.522 Å), d: C18–H18B···O7, d(H···O) = 2.575 Å), e: C17–H17B···S3, d(H···S) = 2.849 Å, π···π, d(π···π) = 3.676(8) Å)

A ORTEP style drawings (50% probability level) of the molecular structures showing the corresponding the inter-ring dihedral angles between the planes in C6. B The intermolecular C–H···Cl/Br/O hydrogen bonding and the intermolecular π···π stacking interactions in C6 (a: C17–H17···Br2, d(H···Br) = 2.850 Å, b: C31–H31···Cl1, d(H···Cl) = 2.846 Å, c: C36–H36B···O2, d(H···O) = 2.602 Å), d: C35–H35B···O2, d(H···O) = 2.583 Å), π···π, d(π···π) = 3.761(7) Å)
CDFT based analysis
Calculated quantum chemical descriptors for L1, L2, L3, C1, C2 and C3 molecules are given in Table 2. Figures 4 and 5 visually present the optimized structure, HOMO and LUMO images of the ligands and complexes, respectively.

The optimized structure, HOMO and LUMO images of the ligands

The optimized structure, HOMO and LUMO images of the complexes
Many theoretical studies presenting the relationships between Conceptual Density Functional Theory based parameters and the biological activity and anticancer effects of the molecules are available in the literature (Gok et al. 2023; Rajkumar et al. 2024). Electronegativity is known as the electron withdrawal power of chemical systems (Kaya and Kaya 2015b). On the other hand, chemical hardness concept reflecting the resistance towards electron cloud polarization of the chemical systems provides useful clues about the stable or reactive behaviors of the compounds (Kaya and Kaya 2015a, c). In the Hard and Soft Acid–Base classification of Pearson (1963), soft chemicals are reported as chemical systems with high polarization ability. Such chemical systems give their electrons easily to another chemical system. According to Maximum Hardness Principle (Kaya et al. 2022), hard chemical systems are highly stable, while soft chemical systems exhibit high reactivity. As a result of the complexation of ligands (L1, L2, L3) with Mo(VI), chemical species (C1, C2, C3) with lower hardness values were formed. As noted in the previously published papers, soft chemical systems show great antioxidant and anticancer activities. In light of the computed chemical hardness values for the studied ligands and their Mo(VI) complexes, it can be said that all complexes exhibit higher anticancer and antioxidant activity than their ligands. C2 and C3 complexes are the most reactive ones among the chemical systems studied. Additionally, it should be noted that chemical hardness and electronegativity values of C2 and C3 complexes are smaller than those of the other chemical systems. For that reason, anticancer and antioxidant activities should be higher than those of the other chemical systems. This comment made based on the results of DFT computations is in good agreement with both experimental cancer analysis studies and docking scores calculated via Molecular Docking analysis. The Minimum Electrophilicity Principle introduced by Chamorro, Chattaraj and Fuentealba (Chamorro et al. 2003) states that in stable states, electrophilicity is minimized. Among the studied Mo(VI) complexes, the most stable one is the C1 complex. If so, it can be said that the Minimum Electrophilicity Principle also accurately predicts the high anticancer and antioxidant activity of C2 and C3 complexes.
Lower chemical hardness (η) and higher electronegativity (χ) are linked to lower IC50 values, which means the blocking effects are stronger. It may not make sense, but higher IC50 values are linked to higher electrophilicity indices (ω1, ω2, ω−, ω+). This could be because these traits and biological activity combine in a complex way. EHOMO and ELUMO: Values that are more positive (less negative) are linked to higher IC50. This means that chemicals with more steady electron distributions (higher EHOMO and ELUMO values) have weaker inhibitory effects. These links show that in this dataset, lower chemical hardness and higher electronegativity are good indicators of higher cellular activity. There is also a good connection between the high electrophilicity indices and the IC50, but more research may be needed to fully understand what this association means.
Antioxidant properties
To determine the antioxidant capacities of the compounds, the CUPRAC method was applied. The molar absorption coefficient (Ɛ) was found from the slope of the absorbance versus concentration graphs of each compound. The TEAC coefficient of each compound was obtained by dividing the molar absorption coefficient (Ɛ) by that of Trolox (Table 3). When Table 3 is examined, it is seen that the TEAC values of both ligands and complexes are higher than the value of Trolox (TEACTrolox = 1). This shows that both ligands and complexes have higher antioxidant capacity compared to Trolox. Also, the TEAC values of all complexes were found to be higher than those of their ligands. In other words, the complexes exhibited better antioxidant capacity than their ligands.
When the free radical scavenging activity results are examined, it is seen that all compounds have less activity than Trolox (Table 4). Similar to the antioxidant capacity results, complexes exhibited slightly better activity than their ligands.
It is well known that the hydroxyl groups of the phenolic structure can donate electrons, which increases the antioxidant capacity. In addition, –NH and –SH groups in thiocarbohydrazones contribute to the overall antioxidant capacity (Charlton et al. 2023; Leopoldini et al. 2011). Although the number of –OH and –NH groups in the complexes was less than the ligands, the antioxidant activity of the complexes was found to be higher compared to the ligands. The acidity of the non-deprotonated –NH group in thiocarbohydrazone, the chelate structure formed, and the contributions of molybdenyl oxygens can be shown as the reason for this (Asha and Prathapachandra Kurup 2020; Kaya 2022). Some other published studies indicated similar results (Asha and Prathapachandra Kurup 2020; Eğlence-Bakır et al. 2021; Kaya 2022; Kaya et al. 2018).
Cytotoxicity results
In order to evaluate the cytotoxic activity of the ligands and complexes against C6 cancer cell lines at concentrations of 100, 50, 25, 12.5, and 6.75 µM for 24, 48, and 72 h (Fig. 6), The untreated cells were used as a control. Cell viability was analyzed by the XTT assay, and the results showed that the complexes and the ligands exhibited an inhibitory effect on the proliferation of C6 cell lines in a dose- and time-dependent manner. According to the results, the C3 complex seems to have the best anti-cancer activity as compared to doxorubicin. Essentially all complexes show higher activity than the ligands. It can be said that this result is based on the presence of molybdenyl cation. The results were analyzed by means of cell inhibition expressed as half-maximum inhibitory concentration (IC50) values. IC50 values are listed in Table 5.

Cell viability by different concentrations of ligands and complexes by incubation time of a 24 h, b, 48 h, c 72 h. The control group is nontreated cells while doxorubicin is standard anticancer drug
Molecular docking analysis
Estimating and comprehending protein-small molecule interactions is critical in biology and drug development. This foresight enables scientists to delve deeper into biological processes, laying the framework for pharmacological research and innovation. Scientists acquire important biological insights by precisely anticipating these interactions. They are capable of detecting minute protein-small molecule interactions, exposing the intricate molecular choreography of physiological activities. This foresight assists scientists in identifying therapeutic targets, thus opening up new drug and therapy options. According to the scientific community, this predictive power is critical for understanding biological operations. Predictive modeling of protein-small molecule interactions aids in the comprehension of complicated biological processes such as cellular signaling networks, protein control and disease pathways. It directs the pursuit of novel medical treatments and discoveries (Guclu et al. 2023; Kaya et al. 2023). Accurate prediction of protein-small molecule interactions is crucial for developing drugs. With this accuracy, researchers can choose drug targets as well as improve drug formulations. Governing biological pathways and developing highly tailored medications that fine-tune protein activity to meet therapeutic aims are made possible by scientists' mastery of these interactions.
The developed ligand molecules were thoroughly evaluated to determine their possible interactions with the protein structures, as shown in Fig. 7. The docking scores acquired provided useful information on the binding strengths of these molecules toward the protein. Predicting protein-molecule interactions is critical for understanding diverse biological processes, decoding protein activities and assisting in medication development (Guclu et al. 2023; Kaya et al. 2023). The results show that the synthesized molecules are prone to different interaction types (hydrogen bonding, π–π, π–σ...) with the 4GG7 chains during the docking procedure, significantly contributing to the interaction strength of the molecules toward the protein’s docking site. The results correspond with the very relatively large negative docking score values obtained as evidenced in previous research (Liu et al. 2020; Trott and Olson 2010).

Corresponding 3D (total and close view) and 2D docking poses for interaction of the: C1, C2 and C3 with the pdb id: 4GG7
The molecules have a strong binding attraction for 4GG7, which can be seen in Fig. 7. It shows that the docking score for both drug structures are very good, ranging from − 155 to − 96 kcal/mol. In conclusion, these results not only show that the drug has a good chance of binding to target proteins, but they also give us a better understanding of the complex molecular processes that make it structurally appropriate for protein interaction. This makes it possible for more study and development to happen in the area of pharmaceutical research. It also creates new opportunities.
Conclusion
Ethanol-solvated Mo(VI) solid complexes from asymmetric bisthiocarbohydrazone ligands containing the heterocyclic isatin structure were synthesized for the first time.
Bright red crystals suitable for X-ray analysis were obtained in an ethanol solution containing DMSO and their three-dimensional structures were elucidated. Ethanol in the sixth coordination was replaced by DMSO, which has higher donor properties.
The anticancer potential of ligands and complexes (C1–C3) against the C6 cell line was examined. The complexes were found to exhibit higher anticancer activity than the ligands, with the C3 complex showing the highest activity. At the same time, all compounds have higher total antioxidant capacity than the standard substance Trolox.
Molecular structure studies show that the bonds of solvent molecules such as methanol and ethanol with the molybdenum atom are greatly lengthened (weakened) due to the static trans effect of the multiple bonded oxo ligands of the [MoO2]2+ group. This enables the desolvation of the complexes when another donor solvent is added (Kaya et al. 2015; Sergienko et al. 2020). This may indicate that alcohol solvated complexes will provide the opportunity for exchange with the substrate when used as biocatalysts.
As a result, molybdenum-based compounds with different functions can be developed and designed to have higher anticancer activity than cisplatin (platinum-based compound).
Data availability
The data that has been used is included in supplementary article and manuscript. The datasets generated and/or analysed during the current study are available in the Protein Data Bank (https://www.rcsb.org/pdb/) repository. The crystallographic files for complexes (C4, C5 and C6) can be downloaded from the Cambridge Crystallographic Data Centre (CCDC No: 2280901, 2280902 and 2280903).
References
Abbas SY, Farag AA, Ammar YA, Atrees AA, Mohamed AF, El-Henawy AA (2013) Synthesis, characterization, and antiviral activity of novel fluorinated isatin derivatives. Monatsh Chem 144(11):1725–1733
Adam MSS, Elsawy H, Sedky A, Makhlouf MM (2022) Comparable catalytic and biological behavior of alternative polar dioxo-molybdenum (VI) schiff base hydrazone chelates. J Taiwan Inst Chem Eng 136:104425
Aldulaijan S, Nabil S, Alharthi S, Abdullatif BA, Abdel-Naby AS (2023) Use of a bioresource nanocomposite as a heterogeneous base catalyst for the green synthesis of novel bioactive pyrazoles: antibacterial evaluation using molecular docking. New J Chem 47(28):13367–13377
Apak R, Güçlü K, Özyürek M, Karademir SE (2004) Novel total antioxidant capacity index for dietary polyphenols and vitamins C and E, using their cupric ion reducing capability in the presence of neocuproine: Cuprac method. J Agric Food Chem 52(26):7970–7981
Asha TM, Prathapachandra Kurup MR (2020) An insight into the potent antioxidant activity of a dithiocarbohydrazone appended cis-dioxidomolybdenum(vi) complexes. Appl Organomet Chem 34(9):e5762
Berisha A (2019a) Interactions between the aryldiazonium cations and graphene oxide: a DFT study. J Chem 2019:1–5
Berisha A (2019b) The influence of the grafted aryl groups on the solvation properties of the graphyne and graphdiyne—a MD study. Open Chem 17(1):703–710
Berk Ş, Kaya S, Akkol EK, Bardakçı H (2022) A comprehensive and current review on the role of flavonoids in lung cancer: experimental and theoretical approaches. Phytomedicine 98:153938
Berman H, Henrick K, Nakamura H (2003) Announcing the worldwide protein data bank. Nat Struct Mol Biol 10(12):980–980
Bitencourt-Ferreira G, de Azevedo WF (2019) Molegro virtual docker for docking. Docking Screens for Drug Discovery 2053:149–167
Bonaccorso C, Marzo T, La Mendola D (2020) Biological applications of thiocarbohydrazones and their metal complexes: a perspective review. Pharmaceuticals 13(1):1–19
Brand-Williams W, Cuvelier M-E, Berset C (1995) Use of a free radical method to evaluate antioxidant activity. LWT-Food Sci Technol 28(1):25–30
Bugalia S, Dhayal Y, Sachdeva H, Kumari S, Atal K, Phageria U, Saini P, Gurjar OP (2023) Review on isatin-a remarkable scaffold for designing potential therapeutic complexes and its macrocyclic complexes with transition metals. J Inorg Organomet Polym Mater 33:1782–1801
Burns GR (1968) Metal complexes of thiocarbohydrazide. Inorg Chem 7(2):277–283
Çavuş MS, Yakan H, Başkan C, Muğlu H, Babacan AA (2024) Schiff bases based on thio/carbohydrazide: synthesis, spectroscopic characterization, DFT, antimicrobial, DNA interactions and cytotoxicity studies. J Mol Struct 1304:137655
Ceylan Bİ, Deniz NG, Kahraman S, Ulkuseven B (2015) Cis-dioxomolybdenum(VI) complexes of a new ONN chelating thiosemicarbazidato ligand; synthesis, characterization, crystal, molecular structures and antioxidant activities. Spectrochim Acta A Mol Biomol Spectrosc 141:272–277
Chakraborty D, Chattaraj PK (2021) Conceptual density functional theory based electronic structure principles. Chem Sci 12(18):6264–6279
Chamorro E, Chattaraj PK, Fuentealba P (2003) Variation of the electrophilicity index along the reaction path. J Phys Chem A 107(36):7068–7072
Charlton NC, Mastyugin M, Török B, Török M (2023) Structural features of small molecule antioxidants and strategic modifications to improve potential bioactivity. Molecules 28(3):1057
Chowdhary S, Shalini AA, Kumar V (2022) A mini review on isatin, an anticancer scaffold with potential activities against neglected tropical diseases (NTDS). Pharmaceuticals 15(5):536
Dolomanov OV, Bourhis LJ, Gildea RJ, Howard JAK, Puschmann H (2009) Olex2: a complete structure solution, refinement and analysis program. J Appl Crystallogr 42(2):339–341
Dragancea D, Arion VB, Shova S, Rentschler E, Gerbeleu NV (2005) Azine-bridged octanuclear copper(II) complexes assembled with a one-stranded ditopic thiocarbohydrazone ligand. Angew Chem 117:8152–8156
Eğlence-Bakır S, Sacan O, Şahin M, Yanardag R, Ülküseven B (2019) Dioxomolybdenum(VI) complexes with 3-methoxy salicylidene-n-alkyl substituted thiosemicarbazones. Synthesis, characterization, enzyme inhibition and antioxidant activity. J Mol Struct 1194:35–41
Eğlence-Bakır S, Şahin M, Özyürek M, Ülküseven B (2021) Dioxomolybdenum(VI) complexes with 4-benzyloxysalicylidene-n/s-alkyl thiosemicarbazones: synthesis, structural analysis, antioxidant activity and xanthine oxidase inhibition. Polyhedron 209:115467
El-Shahawi MS, Shanab MMAH, Mostafa MM (2019) Synthesis and structural studies on some dioxomolybdenum(VI) complexes bearing 1-(1-hydroxynaphthalen-2-yl) ethanone moiety. J Chem Educ Res Pract 3(1):1–14
Fayed AM, Elsayed SA, El-Hendawy AM, Mostafa MR (2014) Complexes of cis-dioxomolybdenum(VI) and oxovanadium(IV) with a tridentate ONS donor ligand: synthesis, spectroscopic properties, X-ray crystal structure and catalytic activity. Spectrochim Acta A Mol Biomol Spectrosc 129:293–302
Fernelius WC, Terada K, Bryant BE (1960) Molybdenum(VI) dioxyacetylacetonate. Inorg Synth 6:147–148
Ferraz de Paiva RE, Vieira EG, Rodrigues da Silva D, Wegermann CA, Costa Ferreira AM (2021) Anticancer compounds based on isatin-derivatives: Strategies to ameliorate selectivity and efficiency. Front Mol Biosci 7:627272
Fouad R, Shaaban IA, Ali TE, Assiri MA, Shenouda SS (2021) Co(II), ni(II), cu(II) and cd(II)-thiocarbonohydrazone complexes: spectroscopic, DFT, thermal, and electrical conductivity studies. RSC Adv 11(60):37726–37743
Gabr MT, El-Gohary NS, El-Bendary ER, El-Kerdawy MM, Ni N (2017) Isatin-β-thiocarbohydrazones: microwave-assisted synthesis, antitumor activity and structure-activity relationship. Eur J Med Chem 128:36–44
Gabr MT, El-Gohary NS, El-Bendary ER, Ni N, Shaaban MI, El-Kerdawy MM (2018) Microwave-assisted synthesis, antimicrobial, antiquorum-sensing and cytotoxic activities of a new series of isatin-β-thiocarbohydrazones. Synth Commun 48(22):2899–2911
Gangarapu K, Manda S, Thota S, Yerra R, Karki SS, Balzarini J, De Clercq E, Tokuda H (2012) Microwave assisted synthesis, characterization of some new isatin and thiophene derivatives as cytotoxic and chemopreventive agents. Lett Drug Des Discov 9(10):934–941
Gangarapu K, Manda S, Jallapally A, Thota S, Karki SS, Balzarini J, De Clercq E, Tokuda H (2014) Synthesis of thiocarbohydrazide and carbohydrazide derivatives as possible biologically active agents. Med Chem Res 23(2):1046–1056
Gázquez JL, Cedillo A, Vela A (2007) Electrodonating and electroaccepting powers. J Phys Chem A 111(10):1966–1970
Geary WJ (1971) The use of conductivity measurements in organic solvents for the characterisation of coordination compounds. Coord Chem Rev 7(1):81–122
Gok E, Unal N, Gungor B, Karakus G, Kaya S, Canturk P, Katin KP (2023) Evaluation of the anticancer and biological activities of istaroxime via ex vivo analyses, molecular docking and conceptual density functional theory computations. Molecules 28(22):7458
Gretarsdóttir JM, Bobersky S, Metzler-Nolte N, Suman SG (2016) Cytotoxicity studies of water soluble coordination compounds with a [Mo2O2S2]2+ core. J Inorg Biochem 160:166–171
Guclu G, Tas A, Dincer E, Ucar E, Kaya S, Berisha A, Dural E, Silig Y (2023) Biological evaluation and in silico molecular docking studies of Abies cilicica (antoine & kotschy) carrière) resin. J Mol Struct 1288:135740
Hashim KKM, Manoj E (2023) DNA and BSA binding studies of new Pd(II) bisthiocarbohydrazone complexes: from anticancer drug analogue to anticovid candidates. Inorg Chem Commun 157:111326
Hille R (2013) The molybdenum oxotransferases and related enzymes. Dalton Trans 42(9):3029–3042
Islam N, Kaya S (2018) Conceptual density functional theory and its application in the chemical domain. CRC Press, New Jersey
Joha Z, Yulak F, Öztürk A, Şahin B, Yıldırım Ş (2021) The anticancer effect of cannabinoid 2 agonist l-759,633 on c6 and sh-sy5y cell lines. Turk J Sci Health 2(3):6–13
Juranić Z, Anastasova F, Juranić I, Stanojković T, Radulović S, Vuletić N (1999) Antiproliferative action of isatine-beta-thiocarbohydrazone and N-ethylisatine-beta-thiocarbohydrazone on human PBMC and on two neoplastic cell lines. J Exp Clin Cancer Res 18(3):317–324
Kaya Y (2022) Investigation of spectroscopic, crystallographic, thermal and antioxidant properties of mononuclear dioxomolybdenum(VI) complexes derived from a new symmetric bisthiocarbohydrazone. Polyhedron 227:116151
Kaya S, Kaya C (2015a) A new method for calculation of molecular hardness: a theoretical study. Comput Theor Chem 1060:66–70
Kaya S, Kaya C (2015b) A new equation based on ionization energies and electron affinities of atoms for calculating of group electronegativity. Comput Theor Chem 1052:42–46
Kaya S, Kaya C (2015c) A new equation for calculation of chemical hardness of groups and molecules. Mol Phys 113(11):1311–1319
Kaya Y, Erçağ A, Koca A (2015) Synthesis, structures, electrochemical studies and antioxidant activities of cis-dioxomolybdenum(VI) complexes of the new bisthiocarbohydrazones. J Mol Struct 1102:117–126
Kaya S, Kaya C, Islam N (2016) Maximum hardness and minimum polarizability principles through lattice energies of ionic compounds. Phys B 485:60–66
Kaya Y, Erçağ A, Kaya K (2018) Synthesis, characterization and antioxidant activities of dioxomolybdenum(VI) complexes of new schiff bases derived from substituted benzophenones. J Coord Chem 71(20):3364–3380
Kaya Y, Erçağ A, Uğuz Ö, Koca A, Zorlu Y, Hacıoğlu M, Birteksöz Tan AS (2021) New asymmetric bisthiocarbohydrazones and their mixed ligand nickel(II) complexes: synthesis, characterization, crystal structure, electrochemical-spectroelectrochemical property, antimicrobial and antioxidant activity. Polyhedron 207:115372
Kaya S, Robles-Navarro A, Mejía E, Gómez T, Cardenas C (2022) On the prediction of lattice energy with the fukui potential: some supports on hardness maximization in inorganic solids. J Phys Chem A 126(27):4507–4516
Kaya Y, Kaya S, Berisha A, Erçağ A (2023) Cyclocondensation of 3,4-diaminobenzophenone with glyoxal: synthesis, X-ray structure, density functional theory calculation and molecular docking studies. J Mol Struct 1291:135973
Klamt A (2005) Cosmo-rs: from quantum chemistry to fluid phase thermodynamics and drug design. Elsevier, Amsterdam
Klamt A (2018) The COSMO and COSMO-RS solvation models. Wires Comput Mol Sci 8(1):e1338
Koopmans T (1934) Über die zuordnung von wellenfunktionen und eigenwerten zu den einzelnen elektronen eines atoms. Physica 1(1–6):104–113
Lebkiri I, Abbou B, Hsissou R, Safi Z, Sadiku M, Berisha A, El Amri A, Essaadaoui Y, Kadiri L, Lebkiri A, Rifi EH (2023) Investigation of the anionic polyacrylamide as a potential adsorbent of crystal violet dye from aqueous solution: equilibrium, kinetic, thermodynamic, DFT, MC and MD approaches. J Mol Liq 372:121220
Leopoldini M, Russo N, Toscano M (2011) The molecular basis of working mechanism of natural polyphenolic antioxidants. Food Chem 125(2):288–306
Liu Y, Grimm M, Dai W-T, Hou M-C, Xiao Z-X, Cao Y (2020) Cb-dock: a web server for cavity detection-guided protein–ligand blind docking. Acta Pharmacol Sin 41(1):138–144
Majumdar A, Sarkar S (2011) Bioinorganic chemistry of molybdenum and tungsten enzymes: a structural-functional modeling approach. Coord Chem Rev 255(9–10):1039–1054
Muğlu H, Yakan H, Misbah AGA, Çavuş MS, Bakır TK (2021) Synthesis, structure characterization and quantum chemical study on relationship between structure and antioxidant properties of novel Schiff bases bearing (thio)/carbohydrazones. Res Chem Intermed 47:4985–5005
Muğlu H, Sönmez F, Çavuş MS, Kurt BZ, Yakan H (2023) New Schiff bases based on isatin and (thio)/carbohydrazone: preparation, experimental–theoretical spectroscopic characterization, and DFT approach to antioxidant characteristics. Res Chem Intermed 49(4):1463–1484
Nair MLH, Thankamani D (2009) Synthesis and characterisation of oxomolybdenum(V) and dioxomolybdenum(VI) complexes with Schiff base derived from isonicotinoylhydrazide. Indian J Chem 48A:1212–1248
Odularu AT, Ajibade PA, Mbese JZ (2019) Impact of molybdenum compounds as anticancer agents. Bioinorg Chem Appl 2019:1–9
Parr RG, von Szentpály L, Liu S (1999) Electrophilicity index. J Am Chem Soc 121(9):1922–1924
Parsekar SU, Paliwal K, Haldar P, Antharjanam PKS, Kumar M (2022) Synthesis, characterization, crystal structure, DNA and HSA interactions, and anticancer activity of a mononuclear cu(II) complex with a Schiff base ligand containing a thiadiazoline moiety. ACS Omega 7(3):2881–2896
Pearson RG (1963) Hard and soft acids and bases. J Am Chem Soc 85(22):3533–3539
Rajkumar K, Gokulakrishnan V, Anand S, Durga R (2024) Spectroscopic, quantum computational, topological, Fukui functions and molecular docking analysis on a potential anti-cancer molecule nicotinamide by DFT method. J Mol Struct 1300:137216
Rakshit S, Palit D, Hazari SKS, Rabi S, Roy TG, Olbrich F, Rehder D (2016) Synthesis, characterization and biomedical activities of molybdenum complexes of tridentate Schiff base ligands. Crystal and molecular structure of [MoO2(L10)(dmso)] and [MoO2(L11)(dmso)]. Polyhedron 117:224–230
Rana A, Dinda R, Sengupta P, Ghosh S, Falvello LR (2002) Synthesis, characterisation and crystal structure of cis-dioxomolybdenum(VI) complexes of some potentially pentadentate but functionally tridentate (ONS) donor ligands. Polyhedron 21(9–10):1023–1030
Rout PK, Roy T, Banerjee DR, Adhikari U, Sukul D, Ghosal S (2023) Theoretical investigation on the adsorption of phospho-tyrosine derivatives over fe (1 1 0) surface in 1 m hcl medium. Trans Indian Inst Met 1–7
Roy S, Saswati LS, Dhaka S, Maurya MR, Acharyya R, Eagle C, Dinda R (2018) Synthesis, structural studies and catalytic activity of a series of dioxidomolybdenum(VI)-thiosemicarbazone complexes. Inorg Chim Acta 474:134–143
Saraswati K, Kant R (2013) Synthesis, characterization and biological activity of some molybdenum(VI) complexes. Der Pharma Chemica 5(4):347–356
Sathisha MP, Revankar VK, Pai KSR (2008) Synthesis, structure, electrochemistry, and spectral characterization of bis-isatin thiocarbohydrazone metal complexes and their antitumor activity against Ehrlich ascites carcinoma in swiss albino mice. Met-Based Drugs 2008:1–11
Sergienko VS, Abramenko VL, Surazhskaya MD (2020) Intracomplex dioxomolybdenum(vi) compounds with alcoholimines of aromatic o-hydroxyaldehydes. Crystal structure of 2-hydroxynaphthylidene monoethanolimine (h2l) and solvated complex [Moo2(l)·C5H5N]. Russ J Inorg Chem 65(4):495–501
Sheldrick GM (2015a) Shelxt–integrated space-group and crystal-structure determination. Acta Crystallogr Sect A Found Adv 71(1):3–8
Sheldrick GM (2015b) Crystal structure refinement with shelxl. Acta Crystallogr Sect C Struct Chem 71(1):3–8
Srividya L, Reddy ARN (2017) Antidiabetic activity of 1-(4-chlorobenzylidene)-5-(2-oxoindolin-3-ylidene) thiocarbohydrazone in chick model. Asian J Biol Sci 10(4):126–129
Tejasree C, Kiran G, Rajyalakshmi G, Reddy ARN (2014) Antidiabetic activity of 1-(4-(dimethylamino)benzylidene)-5-(2-oxo indolin-3-ylidene)thiocarbohydrazone in rats. Int J Pharm Sci Res 5(7):2738
Trott O, Olson AJ (2010) AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem 31(2):455–461
von Szentpály LS, Kaya S, Karakuş N (2020) Why and when is electrophilicity minimized? New theorems and guiding rules. J Phys Chem A 124(51):10897–10908
Wu K, Ai J, Liu Q, Chen T, Zhao A, Peng X, Wang Y, Ji Y, Yao Q, Xu Y, Geng M, Zhang A (2012) Multisubstituted quinoxalines and pyrido[2,3-d]pyrimidines: synthesis and SAR study as tyrosine kinase c-met inhibitors. Bioorg Med Chem Lett 22(20):6368–6372
Yakan H, Çakmak Ş, Buruk O, Veyisoğlu A, Muğlu H, Türköz KN (2022) New 5-methylisatin including thiocarbohydrazones: preparation, structure elucidation and antimicrobial activity. Res Chem Intermed 48(10):4331–4345
Funding
This work was supported by the Scientific Research Projects Coordination Unit of Istanbul University-Cerrahpaşa (project number: FBA-2017-25859).
Author information
Authors and Affiliations
Contributions
YK: Data curation, Investigation, Visualization, Writing-review & editing. AE: Conceptualization, Project administration, Supervision, Writing-original draft, Writing-review & editing. SK: Data curation, Software, Visualization, Writing-review & editing. AB: Data curation, Software, Visualization, Writing-review & editing. BA: Investigation, Data curation. YZ: Data curation, Software, Visualization, Writing-review & editing.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflicts of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Kaya, Y., Erçağ, A., Kaya, S. et al. New solvated Mo(VI) complexes of isatin based asymmetric bisthiocarbohydrazones as potent bioactive agent: synthesis, DFT-molecular docking studies, biological activity evaluation and crystal structures. Biometals (2024). https://doi.org/10.1007/s10534-024-00633-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10534-024-00633-x