
Abstract
Iron is an essential inorganic element for various cellular events. It is directly associated with cell proliferation and growth; therefore, it is expected that iron metabolism is altered in tumor cells which usually have rapid growth rates. The studies on iron metabolism of tumor cells have shown that tumor cells necessitated higher concentrations of iron and the genes of iron uptake proteins were highly over-expressed. However, there are limited number of studies on overall iron metabolism in drug-resistant tumor cells. In this article, we evaluated the studies reporting the relationship between drug resistance and iron metabolism and the utilization of this knowledge for the reversal of drug resistance. Also, the studies on iron-related cell death mechanism, ferroptosis, and its relation to drug resistance were reviewed. We focus on the importance of iron metabolism in drug-resistant cancer cells and how alterations in iron metabolism participate in drug-resistant phenotype.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Iron is an inorganic element which is critical for cell proliferation and growth by incorporating into iron- or heme-containing enzymes. Those enzymes are involved in respiratory complexes, DNA synthesis, cell cycle and detoxification processes. Hence, iron is essential in terms of cell replication, cellular metabolism and growth. Beside these effects, iron can create reactive oxygen species (ROS) by participating in Fenton reaction where hydroxyl radical is produced. ROS can damage DNA and be mutagenic. Thus, iron is not only fundamental for cell survival but it can be related to carcinogenesis (Torti and Torti 2013a, b).
Several studies in the literature relating iron metabolism and cancer revealed that iron is highly demanded by cancer cells. It can facilitate the formation and metastasis of tumor cells, and this detrimental effect can be overcome by iron chelators (Richardson et al. 2009; Torti and Torti 2011, 2013a, b). However, the role of iron metabolism in resistance to chemotherapeutic agents in cancer has not been fully understood.
Drug resistance describes different mechanisms of tumor cells to avoid the cytotoxic effects of anticancer agents with little to no common similarity in terms of structure, function and therapeutic target. Drug-resistant phenotype shows itself with increased insensitivity to chemotherapeutics, leading to decreased success of cancer treatment (Housman et al. 2014). Longley and Johnston stated more than 90% of cancer patients with metastatic tumors are believed to experience failure in chemotherapy due to multidrug resistance (Longley and Johnston 2005). The greater comprehension of cellular mechanisms that drive cancer cells to become drug-resistant will remarkably increase the clinical success of chemotherapy.
Drug resistance, either intrinsic or acquired, develops through alterations in various cellular mechanisms. The cumulative effect of these alterations provides drug-resistant cells with a survival advantage over drug-sensitive ones, leading to formation of drug-resistant tumors. Several studies demonstrated that when over-expressed, ATP-binding cassette (ABC) transporter family proteins pump a wide range of anticancer drugs out of the cell, preventing intracellular drug concentration to reach cytotoxic levels. Apart from the ATP-driven efflux, structural changes in plasma membrane lower membrane permeability; thus, decrease drug influx. Increased activity of cellular detoxification, altered drug targets, enhanced DNA repair, alkaline shift of cellular pH, evasion of apoptosis and increased pro-survival signaling are of importance in development of drug-resistant phenotype (Krishna and Mayer 2000; Lage 2003; Leonessa and Clarke 2003; Longley and Johnston 2005; Simon and Schindler 1994).
In the present review, we evaluated the studies that combine iron metabolism with drug resistance in cancer. We also focused on the strategies to utilize iron metabolism in order to overcome drug resistance. Finally, we compiled the ferroptosis studies associating with the drug resistance in cancer.
Iron metabolism and drug resistance in cancer cells
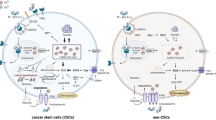
Cellular iron metabolism comprises of three major processes: iron uptake, storage and export (Fig. 1). Firstly, to uptake iron inside the cell, transferrin (Tf) binds free iron (Fe(III)) in circulation and Tf-Fe conjugate then binds to transferrin receptor (TfR) on cell membrane. Tf-Fe-TfR conjugate is internalized via clathrin-dependent endocytosis. Cytoplasmic iron is stored by ferritin heteropolymers which consist of heavy (FtH) and light (FtL) chains. Finally, excess iron is exported by ferroportin (SLC40A1; Hentze et al. 2004).

Cellular iron metabolism. Plasma iron (Fe3+) is captured by transferrin (Tf) in the circulation. Binding of Tf to its receptor (TfR) on the cell membrane activates the receptor and the receptor is internalized in a clathrin-dependent manner. Inside the endosome, Fe3+ is oxidized to Fe2+. Next, Fe2+ is gathered in cytoplasm as labile iron pool and stored in ferritin consisting of heavy chain (FtH) and light chain (FtL). Excess Fe2+ is exported by ferroportin which is regulated by a hormone, hepcidin
In this section, we will basically focus on how expression patterns of these proteins participating in iron uptake, storage and export as well as intracellular free iron levels change in drug-resistant cancer cells compared to their drug-sensitive counterparts. We summarized the altered expression levels of iron metabolism proteins in drug-resistant cell lines in Table 1 and the differences between expression levels of these proteins are given in Fig. 2.

Alterations in iron metabolism due to resistance to anti-cancer drugs. TfR, FtL, FtH, hepcidin and ferroportin are over-expressed (red arrows indicate the over-expression) in drug-resistant cancer cell with minor exceptions. Also, FtL and FtH translocate to nucleus to prevent DNA damage as indicated by red dashed arrows. (Color figure online)
Transferrin receptor and drug resistance
Transferrin receptor (TfR) is a membrane glycoprotein which has a role in importing iron via conjugation to a plasma glycoprotein, transferrin. TfR is a homodimer with 180 kDa molecular weight. Each monomer contains a C-terminal domain that binds transferrin, a single-pass transmembrane domain and a short N-terminal domain. There are four glycosylation sites in C-terminal and this modification is crucial for activity of the receptor. The transcription of TfR gene is regulated by intracellular iron concentration via binding of iron regulatory proteins (IRPs) to the iron response elements (IREs) on the 5′ untranslated region of TfR transcript (Ponka and Lok 1999; Daniels et al. 2006).
Barabas and Faulk have shown that TfR expression was up-regulated in doxorubicin-resistant human chronic myelogenous leukemia cells (K562) and promyclocytic leukemia cells (HL60). Verapamil, a well-known P-glycoprotein inhibitor and a calcium channel blocker, decreased IC50 value of doxorubicin in drug-resistant HL60 cells, which did not over-express P-glycoprotein, in comparison to drug-sensitive cells, by down regulating TfR. TfR can be up-regulated by growth hormones, such as insulin-like growth factor which decreases the rate of receptor endocytosis and epidermal growth factor that increases rate of receptor exocytosis (Davies et al. 1987). This induction was shown to be triggered by calcium channels (Neckers 1991) and the blockade of such mechanism could re-sensitize drug-resistant cells (Cornwell et al. 1987). This may explain the association of TfR with drug resistance through the function of calcium-sensitive receptors (Barabas and Faulk 1993).
A proteomic study to identify membrane protein differences between drug-sensitive and mitoxantrone (MXR)-resistant human breast adenocarcinoma MCF7 cell lines revealed that TfR was highly expressed in MXR-resistant cells while not detected in drug-sensitive parents (Rahbar and Fenselau 2005). Similarly, acquired tamoxifen- and Faslodex-resistant MCF7 cells displayed elevated TfR expression at both mRNA and protein levels (Habashy et al. 2010).
Gallium is a chemical element used as a chemotherapeutic agent, particularly in bladder cancer and lymphoma treatment (Chitambar 2004). Gallium resistance occurs through a unique pathway involving iron metabolism. Because of the fact that gallium has a similar chemistry in terms of ionic radii and bonding to iron, gallium remarkably interferes with iron metabolism (Davies et al. 2006); it can easily conjugate to transferrin and be internalized by TfR-mediated endocytosis. Through this competitive binding to transferrin, gallium inhibits cellular iron uptake and incorporation, and blocks DNA synthesis (Chitambar and Seligman 1986; Chitambar and Zivkovic 1987; Harris and Pecoraro 1983; Larson et al. 1980). In gallium-resistant HL60 cells, TfR was found to be up-regulated, although surprisingly gallium uptake was not increased by high TfR expression compared to drug-sensitive cells. Gallium treatment caused arrest in cellular proliferation in drug-susceptible parents by inhibiting iron uptake whereas drug-resistant HL60 cells continued normal proliferation. Gallium-resistant cells had rapidly up-taken exogenous iron as a result of high levels of TfR on plasma membrane; thus, drug-resistant HL60 cells overcame the effect of gallium by enhanced incorporation of iron (Chitambar et al. 1990). In contrast, Davies et al. reported TfR expression was down-regulated in gallium-resistant HL60 cells and as a result, iron uptake was reduced (Davies et al. 2006). Similarly, in gallium-resistant human leukemic CCRF-CEM cells, TfR expression, and subsequently the iron uptake, was found to be decreased (Chitambar and Wereley 1997).
Chekhun et al. used doxorubicin- and cisplatin-resistant MCF7 cells to investigate differences in expressions of iron metabolism-related proteins. Drug-resistant cells exhibited higher TfR1 expression and iron uptake than drug-sensitive parents. However, intracellular unincorporated Fe(III) levels did not follow the same pattern; in cisplatin-resistant MCF7 cells, free iron levels were 2 times higher than those of drug-sensitive cells whereas there was twofold decrease in doxorubicin-resistant cells (Chekhun et al. 2013, 2017).
Ferritin and drug resistance
Ferritin is an intracellular protein which is responsible for iron sequestration, storage and release. It is composed of two subunits; heavy chain and light chain. These two chains self-assemble and form hetero-24mers in which the ratio of chains is tissue-dependent. Ferritin light chain (FtL) is responsible for mineral nucleation due to its greater negative charge. Ferritin heavy chain (FtH) is involved in nucleation process and also displays ferroxidase activity (Broxmeyer et al. 1991; Koorts and Viljoen 2007; Uchida et al. 2010). The transfection of FtH has been shown to trigger acquired multidrug resistance (MDR) in murine erythroid leukemia (MEL) cells. Epsztejn et al. revealed expression levels of MDR1 and FtH at both mRNA and protein levels were directly proportional. FtH-over-expressing cells were three-to-seven fold resistant to hydrophobic cytotoxic drugs, colchicine and vinblastine, and remarkably affected by P-glycoprotein blockers and MDR reversal agents, cyclosporine and verapamil. The fact that MDR blockers significantly reduced the cellular growth in FtH-over-expressing cells indicates the direct correlation between FtH and multidrug resistance in those cells (Epsztejn et al. 1999).
In gallium-resistant CCRF-CEM cells, heavy chain ferritin mRNA levels were shown to be increased; nevertheless, total ferritin content was lower. The excess transferrin treatment sensitized resistant cells to gallium, implying that gallium resistance is closely linked to iron metabolism (Chitambar and Wereley 1997).
Checkhun et al. proposed the expression of FtH as cell-specific; up-regulated in cisplatin-resistant MCF7 cells but down-regulated in doxorubicin-resistant ones. On the contrary to FtH, FtL was found to be up-regulated in both drug-resistant MCF7 sub-lines; however, sub-cellular localization of FtL differed between drug-sensitive and -resistant cells. As reviewed by Alkhateeb and Connor, nuclear ferritin protects DNA from DNA damage-inducing agents (Alkhateeb and Connor 2010). FtL was cytoplasmic in drug-sensitive parents whilst it was mostly nuclear in drug-resistant counterparts (Chekhun et al. 2013).
FtH expression was also shown to be over-expressed in cisplatin-resistant gastric cancer cells, SNU-620R-CIS/2000, SNU-638R-CIS/400 and SNU-668RCIS/400 in a microarray-based study (Kang et al. 2004). Liu et al. showed that 1,3-bis(2-chloroethyl)-1-nitrosurea (BCNU), which is a DNA-alkaylating agent used in brain tumor therapy (Madajewicz et al. 1981), caused supercoiling of plasmid DNA (pUC19); however, exogenous addition of FtH reversed supercoiling and re-linearized DNA. The change in DNA structure suggests that FtH protected DNA from any damage caused by chemotherapeutics and the presence of FtH in nucleus resulted in decreased sensitivity against anti-cancer agents (Liu et al. 2011).
Bortezomib is a 26S proteasome inhibitor that has an effect on the regulation of various signaling pathways (Chauhan et al. 2005). Campanella et al. reported that bortezomib resistance in multiple myeloma cell lines was correlated with FtH levels. Even though iron supplementation triggered generation of ROS in drug-sensitive cells (MM.1S and KMS-18), such effect was not observed in resistant ones (U266 and RPMI-8226). Down-regulation of FtH in U266 and RPMI-8226 cell lines successfully sensitized the cells to bortezomib (Campanella et al. 2013).
FtH and FtL mRNAs contain iron response elements in their 5’ UTR. These regions were shown to form hairpin loops and impact the regulation of protein synthesis by interacting with other proteins and small molecules (Hentze et al. 2004; Oliveira et al. 1993). The anthracyclines, doxorubicin and daunorubicin, were found to interact with 5’ UTRs of FtH and FtL mRNA (Canzoneri and Oyelere 2008).
Hereditary hemochromatosis protein and drug resistance
Hereditary hemochromatosis protein (HFE) is an atypical major histocompatibility class I molecule that acts as an iron sensor by interacting with TfR1 and TfR2 and regulates cellular iron uptake and whole body iron homeostasis (Goswami and Andrews 2006). Davies et al. revealed a novel mechanism in which HFE formed a secondary complex with transferrin and TfR in gallium-resistant HL60 cells; however, such a complex was absent in drug-sensitive parents. Since HFE competes with transferrin to bind to TfR, it limits the uptake of both iron and gallium in resistant cells (Davies et al. 2006). Moreover, C282Y mutation in HFE gene was shown to be related with resistance to Temodar, geldanamycin, doxorubicin and gamma-radiation in human neuroblastoma (SH-SH5Y) and glioma cells (Lee et al. 2011a, b).
Hepcidin and drug resistance
Hepcidin is a hormone that regulates iron metabolism. It has three isoforms containing 20, 22 and 25 amino acids. The connection between hepcidin and iron metabolism was revealed by iron-overloading studies where hepcidin was over-expressed in hepatocytes. Hepcidin-defficient mice were also shown to have iron over-load in pancreas and liver and hepcidin-overexpressing mice displayed iron deficiency and microcytic hypochromic anemia. By these studies, hepcidin was proposed as a controller of iron transport (Reviewed by Ganz 2003; Nicolas et al. 2002). Yalovenko et al. determined that hepcidin levels were 1.5–2 times higher in doxorubicin- and cisplatin-resistant MCF7 cells. The exogenous administration of hepcidin as well as free iron caused the development of resistance to doxorubicin in rats bearing Walker-256 carcinosarcoma cells (Yalovenko et al. 2016).
Iron efflux, iron accumulation and iron-related proteins, and drug resistance
Watts et al. showed that nitric oxide (NO)-mediated iron efflux together with glutathione was greater in MCF-VP cells which are resistant to VP-16 etoposide and over-express multidrug resistance-associated protein 1 (MRP1) compared to wild-type cells (Watts et al. 2006). Similarly, a multidrug resistance-related protein, breast cancer resistance protein (BCRP; ABCG2) was also found to be responsible for the efflux of heme, a complex of iron and protoporphyrin IX (PPIX) and a regulatory molecule affecting gene expression and translation (Krishnamurthy et al. 2007).
Checkhun et al. showed that free iron complexes were elevated in cisplatin-resistant Guerin carcinoma-transplanted rats (Chekhun et al. 2014).
Tumor associated macrophages (TAMs) was shown to trigger IL-6 secretion in breast cancer cell lines, 4T1 and MCF7, in a paracrine loop manner, resulting in an IL-6 rich niche which generates de novo drug resistance. Iron was proved to enhance this positive feedback loop between TAMs and IL-6, which further strengthens resistance. The fact that an iron chelator, desferrioxamine (DFO), could prevent resistance status by interfering with the function of iron (Li et al. 2016) strongly indicates the importance of iron metabolism in drug resistance. Contrary to that study, Yang et al. determined that DFO promoted resistance to BCNU by activating hypoxia-inducible factor 1 (HIF-1) in rat C6 glioma cells (Yang et al. 2004).
Utilization of iron metabolism to overcome the drug-resistance
From now on, we will focus on the studies that utilize iron metabolism to overcome the drug resistance in cancer cell lines. We deliberately eliminated the resistance status of the drug-resistant cancer cells to iron-related drugs, mainly iron chelators, and were only interested in the restoring sensitivity to anti-cancer drugs via manipulation of cellular iron metabolism. Table 2 summarizes fold decreases in IC50 (inhibitory concentration 50) values of anticancer agents upon alterations in iron metabolism. As seen in Table 2, we classified these strategies into three groups which are explained below.
Transferrin receptor-targeted strategies
Transferrin-conjugated drug studies to overcome the drug resistance were partly reviewed by different research groups (Daniels et al. 2012; Head et al. 1997; Li and Qian 2002; Li et al. 2002; Qian et al. 2002).
Doxorubicin-transferrin conjugate is internalized through a transmembrane mechanism contrary to the function of P-glycoprotein which is active when doxorubicin is administered alone (Li and Qian 2002). Hatano et al. used transferrin-doxorubicin conjugate against doxorubicin-resistant K562 cells and showed that this conjugate reduced the IC50 value from 20 to 3.6 µM (Hatano et al. 1993). Likewise, Fritzer et al. conjugated doxorubicin to transferrin to overcome the multidrug resistance by targeting TfR in multidrug-resistant HeLa contaminant KB cell lines, KB-8-5, KB-C1 and KB-V1. It has been shown that the conjugate completely re-sensitized drug-resistant cells, lowering IC50 values of doxorubicin to that of sensitive ones (Fritzer et al. 1996). In another study by Lai et al., doxorubicin-resistant murine L929 cell line was used to assess the effect of transferrin-doxorubicin conjugate. The transferrin-doxorubicin conjugate mainly localized in cytoplasm while free doxorubicin dispersed through cell membrane, cytoplasm and, nucleus. The conjugation to transferrin did not only provide higher uptake of doxorubicin through TfR but also helps to achieve higher cytoplasmic concentrations of the drug (Lai et al. 1998). The transferrin-doxorubicin conjugate was also shown to lower IC50 of doxorubicin from 7 to 0.035 μM in doxorubicin-resistant human promyelocytic cell line, HL60 (Lubgan et al. 2009). In another study, Fritzer et al. utilized transferrin-doxorubicin conjugate against doxorubicin-resistant HL60 and K562 cell lines and showed that Tf-Dox conjugate significantly decreased the IC50 values in both cell lines (Fritzer et al. 1992).
TfR was targeted by egg-phosphatidylcholine (PC)/cholesterol liposomes encapsulating doxorubicin to overcome MDR in human small cell lung cancer cell line, SBC-3. Transferrin was covalently conjugated to liposomes so as to target TfR-mediated endocytic pathway and overcome P-glycoprotein-mediated drug export. Short-term exposure to TfR-targeted liposome-doxorubicin conjugate increased the toxicity compared to free doxorubicin. Targeted liposomes resulted in high doxorubicin accumulation compared to non-targeted liposome and free doxorubicin in both drug-sensitive and -resistant cells (Kobayashi et al. 2007). In a similar study, Wu et al. designed transferrin-conjugated liposomes co-encapsulating doxorubicin and verapamil (Tf-L-Dox/Ver) to minimize systemic cardiotoxicity of drugs while maximizing cytotoxicity in K562 cell lines. Tf-L-Dox/Ver displayed 5.2 times greater cytotoxicity than non-targeted conjugate and 2.8 times greater cytotoxicity than Tf-L-Dox alone in doxorubicin-resistant K562 cells (Wu et al. 2007). Additionally, doxorubicin-encapsulated anti-TfR antibody-conjugated immunoliposomes were shown to increase intracellular doxorubicin and the duration of intracellular drug accumulation as well as to re-sensitize doxorubicin-resistant cells against the drug (Suzuki et al. 1997). In addition to liposomes, nanoparticles were also used in TfR-targeting strategies. A biodegradable polymer, poly(lactic-co-glycolide) (PLGA) was loaded with paclitaxel and conjugated to transferrin, and assessed in both drug-sensitive and multidrug-resistant MCF7 (MCF7/ADR) cells. Intracellular drug levels, and consequently cell death, were enhanced when treated with Tf-drug conjugate compared to nanoparticles without transferrin conjugation. Such approaches were proposed as new strategies to overcome the multidrug resistance in cancer cells (Sahoo and Labhasetwar 2005).
Artemisinin (ART) is a drug used predominantly in treatment of drug-resistant malaria and also found to be effective in several tumor cells (Klayman 1985; Olliaro et al. 2001; Li et al. 2001). ART only becomes toxic in the presence of Fe2+. Sadava et al. reported human multidrug-resistant small cell-lung cancer (SCLC) cell line H69VP was 10-fold resistant to ART compared to parental H69. Transferrin treatment increased cytotoxicity of ART, achieving complete re-sensitization in H69VP cells. Drug-resistant H69 cell line was shown to have high levels of TfR on cell membrane; therefore, iron uptake was greater than drug-sensitive H69 which, as a result, was not affected by transferrin treatment. They proved the effect of iron by using iron chelator, desferrioxamine mesylate, which reversed the IC50 values of ART with transferrin to ART alone in H69VP while did not affect parental H69 cells. Thus, ART-transferrin co-therapy was offered for the treatment of drug-resistant SCLC (Sadava et al. 2002).
Iron chelators
Iron chelators are complexation agents used to bind free iron and are widely used in treatments of different diseases (Pierre et al. 2002; Sheth 2014; Torti and Torti 2013a). Here, we evaluated the studies that used iron chelators to overcome the drug resistance in cancer.
Whitnall et al. assessed the effect of an iron chelator, di-2-pyridylketone-4,4,-dimethyl3-thiosemicarbazone (Dp44mT), which significantly reversed drug-resistance in etoposide-resistant MCF7 and vinblastine-resistant KB-V1 epidermal carcinoma cells (Whitnall et al. 2006).
In a clinical study, a patient with gemtuzumab ozogamicin-resistant acute monocytic leukemia with a high serum ferritin level was treated with iron chelator, deferasirox. Deferasirox reduced the serum ferritin, eliminating continuous need of blood transfusions and helping complete remission in the patient (Fukushima et al. 2011). Deferasirox was also shown to be effective to overcome the imatinib-resistance in chronic myeloid leukemia cell lines by inducing apoptosis in vitro (Kim et al. 2016).
Iron deprivation by DFO was shown to overcome the multidrug resistance in K562 cell lines by decreasing intracellular iron level and MDR1 expression (Fang et al. 2010).
Ganguly et al. synthesized a novel iron chelator, iron N-(2-hydroxy acetophenone) glycinate (FeNG) that was found to be highly effective in doxorubicin–resistant T lymphoblastic leukemia cell lines, CCRF-CEM. In doxorubicin-resistant cells, FeNG triggered the production of reactive oxygen species (ROS), leading cells to apoptosis (Ganguly et al. 2010). Similarly, FeNG treatment resulted in depletion of intracellular glutathione and increase in effectiveness of doxorubicin in drug-resistant Ehrlich ascites carcinoma cells ex vivo and in vivo (Ganguly et al. 2012).
Beside the iron chelators, several complexes containing iron have been also used to overcome drug resistance in cancer. Lee et al. synthesized a novel iron complex (FeIII(salophene)Cl) which proved remarkably effective in vincristine- and daunorubicin-resistant leukemic Nalm-6 cells (Lee et al. 2011a, b). Another Fe(III)-salophene conjugate was shown to decrease cell viability in a dose-dependent manner by triggering S phase arrest and apoptosis in platinum-resistant ovarian epithelial adenocarcinoma cell lines, SKOV-3 and OVCAR-3 (Lange et al. 2008).
Genetic manipulations
In this strategy, iron metabolism-related proteins are over-expressed and/or silenced to examine the changes in drug resistance upon these alterations.
Liu et al. hypothesized that FtH could protect DNA from damage and the silencing of FtH could increase the sensitivity of glioma cells against the chemotherapeutic agents. It was reported that silencing of FtH decreased lethal dosage (LD) value of BCNU with high active caspase 3 levels in U251 glioma, MCF7 breast adenocarcinoma and malignant peripheral nerve sheath tumor cell lines. Downregulation of FtH also reduced tumor size in drug-treated animal models (Liu et al. 2011). Similarly, in MDA-MB-231 cells, down-regulation of FTH1 by miR-200b brought about increased sensitivity to doxorubicin (Shpyleva et al. 2011).
Chekhun et al. silenced FTL in doxorubicin- and cisplatin-resistant MCF-7 cells by targeting with miR-133a, and showed that downregulation of FTL increased the effectiveness of both drugs on drug-resistant cells (Chekhun et al. 2013).
Ferroptosis and drug resistance
Ferroptosis is a form of non-apoptotic cell death which is dependent on intracellular iron and reactive oxygen species. Intracellular iron triggers reactive oxygen species via the oncogenic Ras-selective lethal small molecule erastin. A small molecule, called ferrostatin-1 and iron chelators were shown to inhibit the ferroptosis (Dixon et al. 2012).
At morphological, biochemical and genetic levels, ferroptosis is distinguishable from other regulated and non-regulated cell death mechanisms. Cells that undergo ferroptosis are rounded up and have dysmorphic mitochondrial phenotype with condensed mitochondrial membrane density (Cao and Dixon 2016; Xie et al. 2016). The induction of cell death in apoptosis-deficient Bax/Bak double knockout mouse embryonic fibroblasts after erastin treatment indicates that ferroptosis in dependent of apoptotic machinery since erastin do not cause apoptotic changes such as chromatin margination, activation of caspases or cleavage of PARP (Dixon et al. 2012). Ferroptosis cannot be attenuated by deletion of Bcl2 family proteins, necroptosis and autophagy inhibitors suggesting that ferroptotic cell death requires different cellular stimulants than other cell death mechanisms (Cao and Dixon 2016). Ferroptosis is triggered by accumulated toxic lipid peroxidation products and iron metabolism-derived reactive oxygen species. This occurs by activation of mitochondrial voltage-dependent anion channels and mitogen-activated protein kinases, enhanced endoplasmic reticulum stress, and inhibition of cystine/glutamate antiporter, unbalancing antioxidant defenses of the cell and resulting in iron-dependent oxidative cell death (Dixon et al. 2012). Misregulated ferroptosis has been shown to be implicated in pathological and physiological conditions (Xie et al. 2016).
Yu et al. used acute myeloid leukemia cell lines to demonstrate the role of ferroptosis inducer, erastin towards activity of the chemotherapeutic drugs, cytarabine and doxorubicin. Treatment with low-dose erastin significantly increased the anticancer activity of cytarabine and doxorubicin in HL60 cells. The enhanced sensitivity to cytarabine and doxorubicin was in a RAS-independent manner given that inhibition of c-Jun and p38, but not ERK, induced resistance to erastin and other anticancer agents (Yu et al. 2015).
Induction of ferroptosis by inhibition of cystine/glutamate antiporter and glutathione peroxidase by cystine/glutamate antiporter inhibitors, erastin and sulfasalazine was shown to overcome cisplatin resistance in head and neck cancer cell lines (AMC-HN3R, -HN4R and –HN9R). Also, high glutamine without cysteine decreased the cell viability even in resistant cell lines, resulting in elevated levels of reactive oxygen species and leading to ferroptosis (Roh et al. 2016). Erastin and sulfasalizine were also assessed to increase the efficiency of Temozolomide (TMZ), an alkylating agent, towards glioblastoma multiforme (GBM) which were proved to be resistant to radio- and chemotherapy. TMZ was shown to increase the expression of xCT, the subunit of glutamate/cysteine transporter system x −c and associating with glutathione (GSH) synthesis and ROS accumulation, via up-regulating NF-E2 related factor 2 (Nrf2) and activating transcription factor 4 (ATF4) transcription factors. Inhibition of xCT by erastin and sulfasalazine decreased the level of GSH and increased the amount of ROS in GBM cell lines, which sensitized the cells towards TMZ via enhanced ferroptosis (Chen et al. 2015). In another study, Nrf2 has been shown to inhibit ferroptosis in cisplatin-resistant head and neck cancer cell lines. Artesunate treatment decreased the cellular GSH levels and increased the ROS formation, which resulted in ferroptosis. Genetic silencing of Nrf2 reversed the inhibition of ferroptosis in these cell lines (Roh et al. 2017).
As an inhibitor of xCT, sulfasalazine also promotes ferroptosis in a similar manner to erastin (Sehm et al. 2016). Therefore, it is not surprising that sulfasalazine has been shown to sensitize colorectal cancer cell lines to cisplatin (Ma et al. 2015); lung adenocarcinoma cell lines and breast cancer cell lines to doxorubicin (Lay et al. 2007; Narang et al. 2007) by triggering ferroptosis.
Sun et al. studied the mechanisms of sorafenib, an inhibitor of multiple oncogenic kinases, in primary tumor hepatocytes, hepatocellular carcinoma lines Huh7, HepaG2 and Hep3b, and in animal models. Sorafenib induced the expression of metallothionein (MT)-1G, which functions in heavy metal detoxification process. Inhibition of MT-1G increased the sensitivity to sorafenib in the cell lines and tumor xenografts models. Furthermore, silencing MT-1G was shown to promote glutathione depletion and lipid peroxidation, triggering ferroptosis in aforementioned cells (Sun et al. 2016).
Conclusions and future perspectives
In this review, we focused on iron metabolism and drug resistance in cancer cells. Resistance to anti-cancer drugs remarkably alters the iron metabolism depending on type of cancer and drugs used. Although drug-resistant phenotype exhibits itself as insensitivity to chemotherapeutics, the genetic alterations leading to drug resistance differ from cell to cell, even in the same cell line that are resistant to different concentrations of the same anticancer agent (Iseri et al. 2012). Since iron is an essential element for cellular proliferation, various drug-resistant cells with rapid proliferation rates, in general, enhance iron uptake and increase intracellular iron level. Iron chelators are proved to be effective to overcome drug resistance in these cells by binding free iron and interfering with overall iron metabolism (Whitnall et al. 2006; Fang et al. 2010; Fukushima et al. 2011; Kim et al. 2016). In other cases, drug resistance seemed to be significantly reversed by supplementation of iron-containing compounds through arrest of cellular growth and, subsequently, enhanced apoptosis (Lee et al. 2011a, b; Lange et al. 2008) although such studies are lack of definition of mechanisms by which these compounds exert their effects. Still, strategies that target iron metabolism definitely increase the efficacy of anti-cancer drugs regardless of the approach. However, more comprehensive studies are still required in this area. As a ROS-dependent cell death mechanism, ferroptosis is greatly linked to drug resistance in cancer. It is known that chemotherapeutic agents trigger elevated ROS formation (Conklin 2004; Arun et al. 2016), however, drug resistant cancer cells develop several mechanisms, particularly increasing GSH levels, to avoid the detrimental effects of ROS (Chen et al. 2015). Any attempt disrupting these mechanisms would inhibit the detoxification processes and could promote ferroptosis. Moreover, accumulation of intracellular free iron would stimulate ROS formation and, thus, ferroptosis (Roh et al. 2017), which would further explain the logic beyond the success of iron treatment in reversal of drug resistance. Furthermore, sulfasalazine, as a ferroptosis-related inhibitor, was shown to be a substrate of breast cancer resistance protein (BCRP/ABCG2; van der Heijden et al. 2004; Tomaru et al. 2013). Elevated levels of BCRP substrates may decrease the rate of drug efflux, which, in turn, triggers ferroptosis and increases the efficiency of drugs. Still, to explore the role of iron metabolism in drug resistance and to overcome the resistance through utilization of iron metabolism seems to be a great challenge.
References
Alkhateeb AA, Connor JR (2010) Nuclear ferritin: a new role for ferritin in cell biology. Biochem Biophys Acta 1800:793–797. doi:10.1016/j.bbagen.2010.03.017
Arun R, Dhivya S, Abraham SK, Premkumar K (2016) Low-dose chemotherapeutic drugs induce reactive oxygen species and initiate apoptosis-mediated genomic instability. Toxicol Res 5:547–556. doi:10.1039/C5TX00391A
Barabas K, Faulk WP (1993) Transferrin receptors associate with drug resistance in cancer cells. Biochem Biophys Res Commun 197:702–708
Broxmeyer HE, Cooper S, Levi S, Arosio P (1991) Mutated recombinant human heavy-chain ferritins and myelosuppression in vitro and in vivo: a link between ferritin ferroxidase activity and biological function. Proc Natl Acad Sci USA 88:770–774
Campanella A, Santambrogio P, Fontana F, Frenquelli M, Cenci S, Marcatti M et al (2013) Iron increases the susceptibility of multiple myeloma cells to bortezomib. Haematologica 98:971–979. doi:10.3324/haematol.2012.074872
Canzoneri JC, Oyelere AK (2008) Interaction of anthracyclines with iron responsive element mRNAs. Nucleic Acids Res 36:6825–6834. doi:10.1093/nar/gkn774
Cao JY, Dixon SJ (2016) Mechanisms of ferroptosis. Cell Mol Life Sci 73:2195–2209
Chauhan D, Hideshima T, Anderson KC (2005) Proteasome inhibition in multiple myeloma: therapeutic Implication. Annu Rev Pharmacol Toxicol 45:465–476
Chekhun VF, Lukyanova NY, Burlaka AP, Bezdenezhnykh NA, Shpyleva SI, Tryndyak VP et al (2013) Iron metabolism disturbances in the MCF-7 human breast cancer cells with acquired resistance to doxorubicin and cisplatin. Int J Oncol 43:1481–1486. doi:10.3892/ijo.2013.2063
Chekhun VF, Lozovska YV, Burlaka AP, Lukyanova NY, Todor IN, Naleskina LA (2014) Peculiarities of antioxidant system and iron metabolism in organism during development of tumor resistance to cisplatin. Exp Oncol 36:196–201
Chekhun VF, Lukyanova NY, Borikun TV, Zadvornyi TV, Mokhir A (2017) Artemisinin modulating effect on human breast cancer cell lines with different sensitivity to cytostatics. Exp Oncol 39:25–29
Chen L, Li X, Liu L, Yu B, Xue Y, Liu Y (2015) Erastin sensitizes glioblastoma cells to temozolomide by restraining xCT and cystathionine-γ-lyase function. Oncol Rep 33:1465–1474
Chitambar CR (2004) Gallium nitrate for the treatment of non-Hodgkin’s lymphoma. Drugs 13:531–541
Chitambar CR, Seligman PA (1986) Effects of different transferrin forms on transferrin receptor expression, iron uptake, and cellular proliferation of human leukemic HL60 cells. J Clin Investig 78:1538–1546
Chitambar CR, Wereley JP (1997) Resistance to the antitumor agent gallium nitrate in human leukemic cells is associated with decreased gallium/iron uptake, increased activity of iron regulatory protein-1, and decreased ferritin production. J Biol Chem 272:12151–12157
Chitambar CR, Zivkovic Z (1987) Uptake of gallium-67 by human leukemic cells: demonstration of transferrin receptor-dependent and transferrin-independent mechanisms. Can Res 47:3929–3934
Chitambar CR, Zivkovic-Gilgenbach Z, Narasimhan J, Antholine WE (1990) Development of drug resistance to gallium nitrate through modulation of cellular iron uptake. J Biol Chem 50:4468–4472
Conklin KA (2004) Chemotherapy-associated oxidative stress: impact on chemotherapeutic effectiveness. Integr Cancer Ther 3:294–300. doi:10.1177/1534735404270335
Cornwell MM, Pastan I, Gottesman MM (1987) Certain calcium channel blockers bind specifically to multidrug-resistant human KB carcinoma membrane vesicles and inhibit drug binding to P-glycoprotein. J Biol Chem 262:2166–2170
Daniels TR, Delgado T, Rodriguez JA, Helguera G, Penichet ML (2006) The transferrin receptor part I: biology and targeting with cytotoxic antibodies for the treatment of cancer. Clin Immunol 121:144–158
Daniels TR, Bernabeu E, Rodríguez JA, Patel S, Kozman M, Chiappetta DA et al (2012) The transferrin receptor and the targeted delivery of therapeutic agents against cancer. Biochem Biophys Acta 1820:291–317. doi:10.1016/j.bbagen.2011.07.016
Davies RJ, Faucher M, Racaniello LK, Carruthers A, Czech MP (1987) Insulin-like growth hormone factor I and epidermal growth factor regulate the expression of transferin receptors at the cell surface by distinct mechanisms. J Biol Chem 262:13126–13134
Davies NP, Rahmanto YS, Chitambar CR, Richardson DR (2006) Resistance to the antineoplastic agent gallium nitrate results in marked alterations in intracellular iron and gallium trafficking: identification of novel intermediates. J Pharmacol Exp Ther 317:153–162
Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE et al (2012) Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 149:1060–1072. doi:10.1016/j.cell.2012.03.042
Epsztejn S, Glickstein H, Picard V, Slotki IN, Breuer W, Beaumont C, Cabantchik ZI (1999) H-Ferritin subunit overexpression in erythroid cells reduces the oxidative stress response and induces multidrug resistance properties. Blood 94:3593–3603
Fang D, Bao Y, Li X, Liu F, Cai K, Gao J, Liao Q (2010) Effects of iron deprivation on multidrug resistance of leukemic K562 cells. Chemotherapy 56:9–16. doi:10.1159/000287352
Fritzer M, Barabas K, Szüts V, Berczi A, Szekeres T, Faulk WP, Goldenberg H (1992) Cytotoxicity of a transferrin-adriamycin conjugate to anthracycline-resistant cells. Int J Cancer 52:619–623
Fritzer M, Szekeres T, Szüts V, Jarayam HN, Goldenberg H (1996) Cytotoxic effects of a doxorubicin-transferrin conjugate in multidrug-resistant KB cells. Biochem Pharmacol 51:489–493
Fukushima T, Kawabata H, Nakamura T, Iwao H, Nakajima A, Miki M et al (2011) Iron chelation therapy with deferasirox induced complete remission in a patient with chemotherapy-resistant acute monocytic leukemia. Anticancer Res 31:1741–1744
Ganguly A, Basu S, Chakraborty P, Chatterjee S, Sarkar A, Chatterjee M, Choudhuri SK (2010) Targeting mitochondrial cell death pathway to overcome drug resistance with a newly developed iron chelate. PLoS ONE. doi:10.1371/journal.pone.0011253
Ganguly A, Chakraborty P, Banerjee K, Chatterjee S, Basu S, Sarkar A et al (2012) Iron N-(2-hydroxy acetophenone) glycinate (FeNG), a non-toxic glutathione depletor circumvents doxorubicin resistance in Ehrlich ascites carcinoma cells in vivo. Biometals 25:149–163. doi:10.1007/s10534-011-9493-7
Ganz T (2003) Hepcidin, a key regulator of iron metabolism and mediator of anemia of inflammation. Blood 102:783–788
Goswami T, Andrews NC (2006) Hereditary hemochromatosis protein, HFE, interaction with transferrin receptor 2 suggests a molecular mechanism for mammalian iron sensing. J Biol Chem 281:28494–28498
Habashy HO, Powe DG, Staka CM, Rakha EA, Ball G, Green AR et al (2010) Transferrin receptor (CD71) is a marker of poor prognosis in breast cancer and can predict response to tamoxifen. Breast Cancer Res Treat 119:283–293. doi:10.1007/s10549-009-0345-x
Harris WR, Pecoraro VL (1983) Thermodynamic binding constants for gallium transferrin. Biochemistry 22:292–299
Hatano T, Okhawa K, Matsuda M (1993) Cytotoxic effect of the protein-doxorubicin conjugates on the multidrug-resistant human myelogenous leukemia cell line, K562, in vitro. Tumor Biol 14:288–294
Head JF, Wang FEN, Elliott RL (1997) Antineoplastic drugs that interfere with iron metabolism in cancer. Adv Enzyme Regul 37:147–169
Hentze MW, Muckenthaler MU, Andrews NC (2004) Balancing acts: molecular control of mammalian iron metabolism. Cell 117:285–297
Housman G, Byler S, Heerboth S, Lapinska K, Longacre M, Snyder N, Sarkar S (2014) Drug resistance in cancer: an overview. Cancers 6:1769–1792. doi:10.3390/cancers6031769
Iseri OD, Kars MD, Gündüz U (2012) Two different docetaxel resistant MCF-7 sublines exhibited different gene expression pattern. Mol Biol Rep 39:3505–3516. doi:10.1007/s11033-011-1123-5
Kang HC (2004) Identification of genes with differential expression in acquired drug-resistant gastric cancer cells using high-density oligonucleotide microarrays. Clin Cancer Res 10:272–284
Kim DS, Na YJ, Kang MH, Yoon S, Choi CW (2016) Use of deferasirox, an iron chelator, to overcome imatinib resistance of chronic myeloid leukemia cells. Korean J Intern Med 31:357–366
Klayman DL (1985) Qinghaosu (Artemisinin): an antimalarial drug from China. Science 228:1049–1055
Kobayashi T, Ishida T, Okada Y, Ise S, Harashima H, Kiwada H (2007) Effect of transferrin receptor-targeted liposomal doxorubicin in P-glycoprotein-mediated drug resistant tumor cells. Int J Pharm 329:94–102
Koorts M, Viljoen M (2007) Ferritin and ferritin isoforms I: structure-function relationships, synthesis, degradation and secretion. Arch Physiol Biochem 113:30–54
Krishna R, Mayer LD (2000) Multidrug resistance (MDR) in cancer: mechanisms, reversal using modulators of MDR and the role of MDR modulators in influencing the pharmacokinetics of anticancer drugs. Eur J Pharm Sci 11:265–283
Krishnamurthy P, Xie T, Schuetz JD (2007) The role of transporters in cellular heme and porphyrin homeostasis. Pharmacol Ther 114:345–358
Lage H (2003) Drug resistance in breast cancer. Cancer Ther 1:81–91
Lai BT, Gao JP, Lanks KW (1998) Mechanism of action and spectrum of cell lines sensitive to a doxorubicin-transferrin conjugate. Cancer Chemother Pharmacol 41:155–160
Lange TS, Kim KK, Singh RK, Strongin RM, McCourt CK, Brard L (2008) Iron(III)-salophene: an organometallic compound with selective cytotoxic and anti-proliferative properties in platinum-resistant ovarian cancer cells. PLoS ONE 3:1–10. doi:10.1371/journal.pone.0002303
Larson SM, Rasey JS, Allen DR, Nelson NJ, Grunbaum Z, Harp GD, Williams DL (1980) Common pathway for tumor cell uptake of gallium-67 and iron-59 via a transferrin receptor. J Natl Cancer Inst 64:41–53
Lay JD, Hong CC, Huang JS, Yang YY, Pao CY, Liu CH, Lai YP, Lai GM, Cheng AL, Su IJ, Chuang SE (2007) Sulfasalazine suppresses drug resistance and invasiveness of lung adenocarcinoma cells expressing AXL. Cancer Res 67(3878):87
Lee SY, Hille A, Kitanovic I, Jesse P, Henze G, Wölfl S et al (2011a) [FeIII(salophene)Cl], a potent iron salophene complex overcomes multiple drug resistance in lymphoma and leukemia cells. Leuk Res 35:387–393. doi:10.1016/j.leukres.2010.11.007
Lee SY, Liu S, Mitchell RM, Slagle-Webb B, Hong YS, Sheehan JM, Connor JR (2011b) HFE polymorphisms influence the response to chemotherapeutic agents via induction of p16INK4A. Int J Cancer 129:2104–2114. doi:10.1002/ijc.25888
Leonessa F, Clarke R (2003) ATP binding cassette transporters and drug resistance in breast cancer. Endocr Relat Cancer 10:43–73
Li H, Qian ZM (2002) Transferrin/transferrin receptor-mediated drug delivery. Med Res Rev 22:225–250
Li Y, Shan F, Wu JM, Wu GS, Ding J, Xiao D, Yang WY, Atassi G, Léonce S, Caignard DH, Renard P (2001) Novel antitumor artemisinin derivatives targeting G1 phase of the cell cycle. Bioorg Med Chem Lett 11:5–8
Li H, Sun H, Qian ZM (2002) The role of the transferrin-transferrin-receptor system in drug delivery and targeting. Trends Pharmacol Sci 23:206–209
Li J, He K, Liu P, Xu LX (2016) Iron participated in breast cancer chemoresistance by reinforcing IL-6 paracrine loop. Biochem Biophys Res Commun 475:154–160. doi:10.1016/j.bbrc.2016.05.064
Liu X, Madhankumar AB, Slagle-Webb B, Sheehan JM, Surguladze N, Connor JR (2011) Heavy chain ferritin siRNA delivered by cationic liposomes increases sensitivity of cancer cells to chemotherapeutic agents. Can Res 71:2240–2249. doi:10.1158/0008-5472.CAN-10-1375
Longley DB, Johnston PG (2005) Molecular mechanisms of drug resistance. J Pathol 205:275–292
Lubgan D, Jóźwiak Z, Grabenbauer GG, Distel LVR (2009) Doxorubicin-transferrin conjugate selectively overcomes multidrug resistance in leukemia cells. Cell Mol Biol Lett 14:113–127. doi:10.2478/s11658-008-0037-2
Ma MZ, Chen G, Wang P, Lu WH, Zhu CF, Song M, Yang J, Wen S, Xu RH, Hu Y, Huang P (2015) Xc- inhibitor sulfasalazine sensitizes colorectal cancer to cisplatin by a GSH-dependent mechanism. Cancer Lett 368:88–96
Madajewicz S, West CR, Park HC, Ghoorah J, Avellanosa AM, Takita H et al (1981) Phase II Study-Intra-arterial BCNU therapy for metastatic brain tumors. Cancer 47:653–657
Narang VS, Pauletti GM, Gout PW, Buckley DJ, Buckley AR (2007) Sulfasalazine-induced reduction of glutathione levels in breast cancer cells: enhancement of growth-inhibitory activity of Doxorubicin. Chemotherapy 53:210–217. doi:10.1159/000100812
Necker LM (1991) Regulation of transferin receptor expression and control of cell growth. Pathobiology 59:11–18
Nicolas G, Viatte L, Bennoun M, Beaumont C, Kahn A, Vaulont S (2002) Hepcidin, a new iron regulatory peptide. Blood Cells Mol Dis 29:327–335
Oliveira CC, Goossen B, Zanchin NIT, Mccarthy JEG, Hentze MW, Stripecke R (1993) Translational repression by the human iron-regulatory factor (IRF) in saccharomyces cerevisiae. Nucleic Acids Res 21:5316–5322
Olliaro PL, Haynes RK, Meunier B, Yuthavong Y (2001) Possible modes of action of the artemisinin-type compounds. Trends Parasitol 17:122–126
Pierre JL, Fontecave M, Crichton RR (2002) Chemistry for an essential biological process: the reduction of ferric iron. Biometals 15:341–346
Ponka P, Lok CN (1999) The transferrin receptor: role in health and disease. Int J Biochem Cell Biol 31:1111–1137
Qian ZM, Li H, Sun H, Ho K (2002) Targeted drug delivery via the transferrin receptor-mediated endocytosis pathway. Pharmacol Rev 54:561–587
Rahbar AM, Fenselau C (2005) Unbiased examination of changes in plasma membrane proteins in drug resistant cancer cells. J Proteome Res 4:2148–2153
Richardson DR, Kalinowski DS, Lau S, Jansson PJ, Lovejoy DB (2009) Cancer cell iron metabolism and the development of potent iron chelators as anti-tumour agents. Biochem Biophys Acta 1790:702–717. doi:10.1016/j.bbagen.2008.04.003
Roh JL, Kim EH, Jang HJ, Park JY, Shin D (2016) Induction of ferroptotic cell death for overcoming cisplatin resistance of head and neck cancer. Cancer Lett 381:96–103. doi:10.1016/j.canlet.2016.07.035
Roh JL, Kim EH, Jang H, Shin D (2017) Nrf2 inhibition reverses the resistance of cisplatin-resistant head and neck cancer cells to artesunate-induced ferroptosis. Redox Biol 11:254–262
Sadava D, Phillips T, Lin C, Kane SE (2002) Transferrin overcomes drug resistance to artemisinin in human small-cell lung carcinoma cells. Cancer Lett 179:151–156
Sahoo SK, Labhasetwar V (2005) Enhanced antiproliferative activity of transferrin-conjugated paclitaxel-loaded nanoparticles is mediated via sustained intracellular drug retention. Mol Pharm 2:373–383
Sehm T, Fan Z, Ghoochani A, Rauh M, Engelhorn T, Minakaki G, Dorfler A, Klucken J, Buchfelder M, Eyupoglu IY, Savaskan N (2016) Sulfasalazine impacts on ferroptotic cell death and alleviates the tumor microenvironment and glioma-induced brain edema. Oncotarget 7:36021–36033
Sheth S (2014) Iron chelation: an update. Curr Opin Hematol 21:1–7. doi:10.1097/MOH.0000000000000031
Shpyleva SI, Tryndyak VP, Kovalchuk O, Starlard-Davenport A, Chekhun VF, Beland FA, Pogribny IP (2011) Role of ferritin alterations in human breast cancer cells. Breast Cancer Res Treat 126:63–71. doi:10.1007/s10549-010-0849-4
Simon SM, Schindler M (1994) Cell biological mechanisms of multidrug resistance in tumors. Proc Natl Acad Sci USA 91:3497–3504
Sun X, Niu X, Chen R, He W, Chen D, Kang R, Tang D (2016) Metallothionein-1G facilitates sorafenib resistance through inhibition of ferroptosis. Hepatology 64:488–500. doi:10.1002/hep.28574
Suzuki S, Inoue K, Hongoh A, Hashimoto Y, Yamazoe Y (1997) Modulation of doxorubicin resistance in a doxorubicin- resistant human leukaemia cell by an immunoliposome targeting transferring receptor. Br J Cancer 76:83–89
Tomaru A, Morimoto N, Morishita M, Takayama K, Fujita T, Maeda K, Kusuhara H, Sugiyama Y (2013) Studies on the intestinal absorption characteristics of sulfasalazine, a breast cancer resistance protein (BCRP) substrate. Drug Metab Pharmacokinetic 28:71–74
Torti SV, Torti FM (2011) Ironing out cancer. Can Res 71:1511–1514. doi:10.1158/0008-5472.CAN-10-3614
Torti SV, Torti FM (2013a) Cellular iron metabolism in prognosis and therapy of breast cancer. Crit Rev Oncog 18:435–448
Torti SV, Torti FM (2013b) Iron and cancer: more ore to be mined. Nat Rev Cancer 13:342–355. doi:10.1038/nrc3495
Uchida M, Kang S, Reichhardt C, Harlen K, Douglas T (2010) The ferritin superfamily: supramolecular templates for materials synthesis. Biochem Biophys Acta 1800:834–845. doi:10.1016/j.bbagen.2009.12.00
Van der Heijden J, de Jong MC, Dijkmans BAC, Lems WF, Oerlemans R, Kathmann I, Schalkwijk CG, Scheffer GL, Scheper RJ, Jansen G (2004) Development of sulfasalazine resistance in human T cells induces expression of the multidrug resistance transporter ABCG2 (BCRP) and augmented production of TNFa. Ann Rheum Dis 63:138–143. doi:10.1136/ard.2002.005249
Watts RN, Hawkins C, Ponka P, Richardson DR (2006) Nitrogen monoxide (NO)-mediated iron release from cells is linked to NO-induced glutathione efflux via multidrug resistance-associated protein 1. Proc Natl Acad Sci USA 103:7670–7675
Whitnall M, Howard J, Ponka P, Richardson DR (2006) A class of iron chelators with a wide spectrum of potent antitumor activity that overcomes resistance to chemotherapeutics. Proc Natl Acad Sci USA 103:14901–14906
Wu J, Lu Y, Lee A, Pan X, Yang X, Zhao X, Lee RJ (2007) Reversal of multidrug resistance by transferrin conjugated liposomes co-encapsulating doxorubicin and verapamil. J Pharm Pharm Sci 10:350–357
Xie Y, Hou W, Song X, Yu Y, Huang J, Sun X et al (2016) Ferroptosis: process and function. Cell Death Differ 23:369–379. doi:10.1038/cdd.2015
Yalovenko TM, Todor IM, Lukianova NY, Chekhun VF (2016) Hepcidin as a possible marker in determination of malignancy degree and sensitivity of breast cancer cells to cytostatic drugs. Exp Oncol 38:84–88
Yang DI, Chen SD, Yang YT, Ju TC, Xu JM, Hsu CY (2004) Carbamoylating chemoresistance induced by cobalt pretreatment in C6 glioma cells: putative roles of hypoxia-inducible factor-1. Br J Pharmacol 141:988–996. doi:10.1038/sj.bjp.0705687
Yu Y, Xie Y, Cao L, Yang L, Yang M, Lotze MT, Zeh HJ, Kang R, Tang D (2015) The ferroptosis inducer erastin enhances sensitivity of acute myeloid leukemia cells to chemotherapeutic agents. Mol Cell Oncol 2:e1054549. doi:10.1080/23723556.2015.1054549
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Kazan, H.H., Urfali-Mamatoglu, C. & Gunduz, U. Iron metabolism and drug resistance in cancer. Biometals 30, 629–641 (2017). https://doi.org/10.1007/s10534-017-0037-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10534-017-0037-7