
Abstract
The trace element molybdenum (Mo) is essential to a suite of nitrogen (N) cycling processes in ecosystems, but there is limited information on its distribution within soils and relationship to plant and bedrock pools. We examined soil, bedrock, and plant Mo variation across 24 forests spanning wide soil pH gradients on both basaltic and sedimentary lithologies in the Oregon Coast Range. We found that the oxidizable organic fraction of surface mineral soil accounted for an average of 33 % of bulk soil Mo across all sites, followed by 1.4 % associated with reducible Fe, Al, and Mn-oxides, and 1.4 % in exchangeable ion form. Exchangeable Mo was greatest at low pH, and its positive correlation with soil carbon (C) suggests organic matter as the source of readily exchangeable Mo. Molybdenum accumulation integrated over soil profiles to 1 m depth (τMoNb) increased with soil C, indicating that soil organic matter regulates long-term Mo retention and loss from soil. Foliar Mo concentrations displayed no relationship with bulk soil Mo, and were not correlated with organic horizon Mo or soil extractable Mo, suggesting active plant regulation of Mo uptake and/or poor fidelity of extractable pools to bioavailability. We estimate from precipitation sampling that atmospheric deposition supplies, on average, over 10 times more Mo annually than does litterfall to soil. In contrast, bedrock lithology had negligible effects on foliar and soil Mo concentrations and on Mo distribution among soil fractions. We conclude that atmospheric inputs may be a significant source of Mo to forest ecosystems, and that strong Mo retention by soil organic matter limits ecosystem Mo loss via dissolution and leaching pathways.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
The trace metal molybdenum (Mo) is an essential micronutrient in ecosystem cycling of nitrogen (N), carbon (C), and other elements essential to life. Perhaps the most prominent role for Mo is as a co-factor in the nitrogenase enzyme, which can lead to Mo limitation of biological N2 fixation when Mo is in short supply (Silvester 1989; Barron et al. 2008; Wurzburger et al. 2012; Jean et al. 2013). Molybdenum also influences nitrification, nitrate assimilation, and denitrification, and is thus broadly significant in shaping ecosystem N cycling (Stiefel 2002). Nitrogen availability, in turn, can control terrestrial productivity by altering soil chemistry and limiting plant growth and C storage (Vitousek and Howarth 1991; Thomas et al. 2010; Menge 2011). Agricultural research has long emphasized an important role for Mo in the growth and health of both N-fixing and non-fixing crop plants, which has also contributed to understanding of Mo in soil systems (Gupta 1997; Kaiser et al. 2005), but comparatively little work has examined soil Mo availability and cycling in natural ecosystems.
Trace metal concentrations in soil organic (O) horizons are a function of biological recycling and atmospheric inputs. Biologically recycled trace metals reflect the metal concentrations of plant tissue, processes that occur during senescence, and processes that occur during the decomposition of plant litter. Studies examining Mo during foliar senescence have observed that the Mo content of foliage remains constant in some cases (Kraepiel et al. 2015), or increases in others (Tyler 2005). During litter decomposition on the forest floor, Mo concentrations can increase dramatically, even when normalized to the loss of matter through mineralization (Brun et al. 2008). This suggests that atmospheric inputs of Mo, which are rarely measured, may strongly influence the amount of Mo in forest soil O horizons.
The primary factors thought to regulate mineral soil Mo availability are pH and sorption by iron (Fe) oxides. At low pH, Mo becomes strongly adsorbed to Fe, manganese (Mn), and aluminum (Al) oxides (Karimian and Cox 1979; Goldberg and Forster 1998; Goldberg et al. 2002; Xu et al. 2006). At high pH, Mo solubility increases with decreased adsorption to Fe and Mn oxides, which can in turn increase Mo loss as soluble molybdate (MoO4 2−). Oxidation and reduction reactions in the soil can also release Mo from adsorption sites, enhancing Mo bioavailability (Gupta 1997). Molybdenum can also bind to soil organic matter through ligand exchange and specific adsorption, which prevents Mo leaching (Wichard et al. 2009). Other studies have also noted the potential importance of organic matter-Mo associations (e.g. Karimian and Cox 1978; Bibak and Borggard 1994; McBride et al. 2000; Wurzburger et al. 2012), although natural organic matter-Mo associations have rarely been quantified. Despite identification of these mechanisms of Mo retention in soil, there are few studies that evaluate their relative importance for overall Mo retention in forest soils. In particular, it is unknown how these mechanisms vary across soils, or the degree to which they influence net Mo retention or loss relative to bedrock, which is needed to interpret constraints on whole-ecosystem Mo balances and bioavailability.
Understanding the relative importance of organic matter in regulating soil Mo behavior is essential for determining the bioavailability of soil Mo to N-fixing organisms, and in turn, the potential N-supply capacity of an ecosystem. If soil organic matter is a dominant control on soil Mo, long-term Mo availability will be tied to pH, organic matter decomposition dynamics and the specific binding mechanisms of Mo to organic matter. On the other hand, if Fe oxide adsorption dominates Mo behavior, Fe oxide surface characteristics and abundance, pH, and redox processes are likely to control Mo bioavailability. Evaluating variation in Mo storage along soil gradients that vary in these key soil properties can help resolve the relative importance of organic matter versus Fe oxide sorption as mechanisms of Mo retention in soils.
Labile phases of Mo may be especially important to ecosystem Mo cycling due to their potential to be taken up by plants and microorganisms. To investigate bioavailability, selective chemical extractions of trace metals are commonly compared to plant tissue concentrations (Kabata-Pendias and Pendias 2001). However, soil chemical extractions targeting labile Mo phases often yield inconsistent results, correlating in some cases to foliar Mo concentrations (Jarrell and Dawson 1978; Lombin 1985; Lang and Kaupenjohann 1998) while showing no relationship in others (Karimian and Cox 1979; Bhella and Dawson 1972; Mortvedt and Anderson 1982; Lang and Kaupenjohann 2000). Similarly, labile Mo abundance is not necessarily correlated with Mo nutrient limitation of heterotrophic soil N2 fixation (Jean et al. 2013). These results suggest that the utility of extractable Mo measurements may remain quite limited because many other soil factors affect Mo bioavailability (Cox and Kamprath 1972; Kubota and Cary 1982). Given that labile Mo phases are likely to exhibit rapid turnover and uptake, longer-term Mo supply from larger Mo pools may be more important for assessing soil Mo dynamics and availability. This may be especially true in forest ecosystems, where trees are long-lived, and where Mo can be recycled annually to soil via plant uptake and litterfall.
We investigated controls on the abundance and distribution of soil Mo at 24 forested sites across the Oregon Coast Range on both basaltic and sedimentary bedrock. These sites occupy a narrow range of climate and current conifer vegetation, yet span wide gradients in 0–10 cm depth soil \({\rm pH}_{(_{{{\text{H}}_{2} {\text{O}}}})}\) (3.9–6.5) and C content (2.5–16.7 %) that reflect historical symbiotic N2 fixation effects on site productivity, decomposition, soil nitrification, acidification, and base cation depletion (Perakis et al. 2006, 2012, 2013). Forest soils of this region also display Mo limitation of heterotrophic N2 fixation (Silvester 1989). We sampled organic and mineral soil, tree foliage, and bedrock at all sites, and quantified surface mineral soil Mo fractions using the following selective chemical extractions: (1) exchangeable Mo (2) reducible Mo associated with short-range-order Fe, Mn, and Al oxides, and (3) oxidizable Mo associated with soil organic matter. Our first objective was to assess the controls on, and the relative importance of exchangeable Mo, reducible Mo, and oxidizable Mo, taking advantage of the gradients in soil properties and contrasting bedrock types. We expected that the amount of reducible Mo would increase at low pH, reflecting pH control of adsorption as a dominant control on overall Mo retention in soil. Our second objective was to investigate controls on Mo mobility by calculating the net loss or gain of Mo relative to parent material inputs and to assess the controls on Mo mobility by comparing the net loss or gain to soil properties. Our third objective was to evaluate the role of biological recycling of soil Mo, through comparison of the potentially bioavailable extractable mineral soil Mo fractions with foliar Mo, and by comparison of foliar Mo with Mo in the soil O horizon. In this comparison, we expected that litterfall fluxes would be larger than atmospheric Mo fluxes, causing foliar Mo concentrations across 24 forested sites with variable foliar Mo to correlate with O horizon soil Mo concentrations, reflecting internal biological recycling of Mo.
Methods
Study site
We examined 24 young Douglas-fir (Pseudotsuga menziesii) forests in the Oregon Coast Range on both sedimentary and basaltic bedrock (Fig. 1; Tables 1, 2). The Oregon Coast Range has a Mediterranean climate regime, characterized by warm, dry summers and cool, wet winters. Mean annual precipitation across our study sites ranges from 180 to 380 cm yr−1 with peak rainfall occurring during the winter and early spring (November–May). Mean annual minimum and maximum temperatures range from 3.97 to 6.02 °C and 13.1 to 16.5 °C across the field area (PRISM Climate Group, Oregon State University). Native vegetation includes late successional Douglas-fir and western hemlock (Tsuga heterophylla), though large areas are currently managed for Douglas-fir timber production on rotation intervals of 50 years or less (Spies et al. 2002).

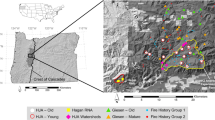
Simplified geologic map, and field sampling locations in the northern Oregon Coast Range, USA. Three sites with filled circles (two sedimentary and one mixed lithology site) indicate the locations of the N-S rainfall collectors. The line of six filled circles along the southern margin of the map indicate the E-W rainfall collector transect
The sites were selected to span a wide gradient in soil N (sedimentary: 0.16–0.86 % N, basaltic: 0.21–0.97 % N, 0–10 cm), accompanied by a wide gradient in soil pH (sedimentary: 6.17–3.90, basaltic: 6.45–4.55, 0–10 cm), and soil C (sedimentary: 2.5–15.7 %, basaltic: 5.0–16.7 %, 0–10 cm) (Table 2). Of the 24 study sites, 11 occurred on sedimentary bedrock, and 11 on basaltic bedrock. The eleven sedimentary sites were established on Eocene-Tertiary sedimentary rock of the Nestucca, Yamhill, and Tyee formations. These rocks include sandstone, siltstone, and mudstone units, with lithic, arkosic micaceous, and basaltic wackes. Quartz, andesine, and lithic fragments are dominant, with trace potassium feldspars, smectite, and calcite (Snavely et al. 1964; McWilliams 1973; Heller et al. 1985). The eleven igneous sites were established on basaltic parent material from the Siletz River Volcanics, Tillamook Volcanics, and Lee’s Falls Formations (Snavely et al. 1968, Snavely and MacLeod 1974). Two additional sites were established on mixtures of the sedimentary and basaltic lithologies.
Field methods
Mineral soil samples were collected in fall of 2012 by removing the O horizon and collecting the 0–10 cm depth with a 6.8 cm diameter corer. Soil at depth was collected with a 4.7 cm diameter slide hammer in 20 cm (or 30 cm for 70–100 cm depth) increments to maximum depth of 100 cm. Soil within the tip was discarded, and each 20 cm section was split equally by height into 10 cm fractions. Three replicate cores from each site were combined in the laboratory into a single sample, and sieved (2 mm) to remove coarse fragments prior to chemical extractions of the field-moist, fine-earth fraction. Samples of O horizon soil were collected as grab samples to the surface of mineral soil in April 2014 as a composite from multiple locations per site (n > 5 per plot). Composite samples were homogenized in the laboratory, and sorted by hand to remove visible mineral soil, rocks, living moss, and sticks larger than 1 cm diameter.
Samples of ‘sun’ foliage from three Douglas-fir trees per site were collected in June 2012 from the 5th whorl of southwest-facing branches, and composited into a single sample prior to analysis. Foliage samples were sorted in the laboratory to separate out first-year needles for analysis, which standardizes biomass compartments across sites when characterizing current tree Mo utilization. Bedrock was collected by portable drill at each site, or by drilling over a nearby outcrop when bedrock depth exceeded the reach of the drilling equipment (>15 m; sites 143, 8, Jenson, and BC1).
Rainwater was collected by event-based sampling using 1.0 L acid-washed plastic bottles with polypropylene funnels covered by 2 mm mesh. First, samples were collected from separate storm events on February 21–26, and April 4–6 2013 at three locations corresponding to soil sampling sites spanning the north–south range of our study sites. Second, rainwater was collected at six locations along an east–west transect across the Coast Range on March 7–14 and October 11–18 of 2014.
Laboratory methods
Mineral soil, O horizon, and foliar samples were oven dried for 48 h. Mineral soil chemistry is reported based on 105 °C dry mass, and O horizon soil chemistry and foliar chemistry are reported based on 65 °C dry mass. Foliar and O horizon soil samples were powdered using a trace metal free agate ball mill, as Mo concentrations can be hampered by Mo contamination introduced during sample processing, particularly with grinding machines using steel or other metallic surfaces (Marks et al. in press). Dried, ground foliage and soil were analyzed for C and N concentrations by Costech ECS 4010 at the USGS Forest and Rangeland Ecosystem Science Center (Corvallis, OR). Foliage was digested using approximately 200 mg of powdered needles in concentrated HNO3 and H2O2. All rainwater samples were acidified with ultra pure HNO3 and evaporated on a hot plate, with concentrated ultrapure HNO3 used to dissolve fine particulates.
Soil exchangeable Al and exchangeable acidity were determined using a 1.0 M potassium chloride (KCl) extraction on ~3 g of field moist soil. Total acidity was determined by titrating ~30 mL of extractant with 0.01 M NaOH to a phenolphthalein endpoint. Aluminum acidity was measured by adding 5 mL of 0.5 M sodium fluoride (NaF) to the titrated extractant and back-titrating with 0.01 M HCl. Soil pH was measured in a 2:1 soil–water mixture (by mass) following a 30 min incubation. Effective cation exchange capacity (ECEC) was calculated using total exchangeable acidity and exchangeable Na, K, Ca, and Mg concentrations. Base saturation was calculated using ECEC and total exchangeable acidity. Hurst’s Redness Rating was calculated using Eq. 1 (Hurst 1977) with color values measured on moist soil samples, where RR is the Hurst’s redness rating, H * is the modified soil hue, V is the soil value, and C is the chroma.
Exchangeable Mo was determined by 1.0 M ammonium (NH4OAc) acetate extraction on ~5 g of field moist soil (Burt 2004; Neunhauser et al. 2001). We extracted reducible Mo, targeting short-range order Fe, Al, and Mn-oxides using a two-step adaptation of the classic Tessier et al. (1979) and BCR (Ure et al. 1993; Rauret et al. 1999) methods. We chose the hydroxylamine hydrochloride extraction over another common method, citrate dithionite, because the citrate dithionite extraction is known to have higher Mo contamination and risk of metal sulfide precipitation. The extraction was performed twice: once using a 0.04 M extraction solution (RED—0.04 M) (Tessier et al. 1979) and once using a 0.1 M extraction solution (RED—0.1 M) (Ure et al. 1993). Two grams of field-moist soil were mixed with 40 mL of hydroxylamine hydrochloride (NH2OH·HCl) in 25 % (v/v) acetic acid (CH3COOH) adjusted to pH 2 with concentrated HNO3. Samples were shaken over night and then heated at 80 °C for 6 h. Two oxidizable pool extractions were performed; one sequential and one non-sequential. The non-sequential extraction (SOM1) was performed on ~2 g of field moist soil. The sequential extraction (SOM2) was performed on rinsed residue from the RED—0.1 M extraction. To oxidize soil organic matter and liberate organically-bound metals, we used a 3-day procedure adapted from the BCR scheme (Ure et al. 1993). Initially, 3 mL 0.02 M ultrapure HNO3 and 5 mL 30 % trace metal grade H2O2 (adjusted to pH 2) were added to 2 g of soil in a 2:1 water-soil slurry. After 12 h, the slurry was heated at 85 °C for 3 additional hours. Hydrogen peroxide was added to the heated samples in 2 mL aliquots every 45 min until effervescence ceased and we observed a visible lightening in the color of the soil. After the slurry had cooled, 5 mL of 3.2 M NH4OAc in 20 % HNO3 was added to prevent re-adsorption of extracted metals to oxide surfaces. After sitting over-night, the slurry was centrifuged at 3000 rpm for 20 min. The supernatant was removed and the remaining soil was rinsed with 75 mL of Milli-Q water (18.2 MΩ). Rinse water was saved with supernatant. For all extractions, supernatant was filtered with a 0.45 µm syringe tip filter prior to preparation for elemental analysis.
Bulk soil chemistry for mineral soil was determined using the ‘residue’ remaining after the exchangeable-pool extraction. This residue was dry-ashed at 500 °C for 12 h, ground, and homogenized. For both mineral soil residues, and bulk O horizon soil samples, approximately 200 mg was microwave digested in a mixture of concentrated ultrapure HNO3, HF, and HCl. Exchangeable Mo concentrations were added to the mineral soil residue Mo concentrations to calculate a bulk-soil concentration. Crushed bedrock samples were powdered using an agate ball mill. Approximately 200 mg of bedrock power was digested using the same procedure as the soil digests. We used data from bulk soil and bedrock digests to calculate total mobility (τMoNb) of bulk soil Mo relative to the index element niobium (Nb), using the elemental mass transfer function:
where τj,w is relative loss or gain of element j from soil with respect to parent material, C j,w is the molar concentration of element j in soil and C j,p is the molar concentrations of element j in bedrock, C Nb,w and C Nb,p are the molar concentrations of the index element in soil and bedrock, respectively (Brimhall and Dietrich 1987; Chadwick et al. 1990). The tau (τ) value allows us to quantify the gain or loss of an element in soil relative to the original concentration of that element in bedrock. We used the immobile element niobium (Nb), which has previously been shown to be the least mobile element in basaltic soils (Kurtz et al. 2000). A tau-value of +1 indicates 100 % gain relative to bedrock, while a value of -1 indicates complete elemental loss relative to bedrock contribution.
All chemical extractions, rock and soil residue digest, and O horizon soil digests were dried-down on a hot plate and re-suspended in ~1 % quartz distilled HNO3 before being analyzed for major and trace elements. Foliar digests were diluted using ~1 % quartz distilled HNO3 and analyzed without being dried down or re-suspended. Trace element concentrations were determined by inductively coupled plasma mass spectrometry (ICP-MS) and major element concentrations were determined by ICP-Optical Emission Spectrometry at the W.M. Keck Collaboratory for Plasma Spectrometry at Oregon State University. For major element chemistry of reducible pool extracts, bedrock, and bulk soil, three USGS rock standards (AGV1 andesite, BCR2 basalt, and G2 granite) were used as calibration standards. Mixed element solution standards were used as calibration standards for minor element chemistry in foliage, bulk soil, bedrock, and reducible pool extracts. Separate plant standards were measured during each analysis for quality control. For major and minor element chemistry on O horizon soil, and major element chemistry on oxidizable-pool extracts, two NIST plant standard reference materials (SRM1515 apple leaf and SRM1570a spinach leaf) were used as calibration standards, along with one International Atomic Energy Agency (IAEA) plant standard (IAEA-336 lichen). Procedural blanks of Mo were always less than 1 %, except in the case of exchangeable and reducible extractions. Because exchangeable and reducible Mo yields were so low, reagent Mo blanks were relatively large, representing, on average, 6 and 30 % of measured concentrations respectively. Oxidizable Mo reagent blanks represented less than 2 % of measured concentrations, and complete digestion blanks were negligible. Reagent blanks have been subtracted from all reported data. For each extraction, 3–4 duplicate samples were separately extracted; these duplicates agreed within 5 % for exchangeable Mo, 16 % for reducible Mo, and 8 % for oxidizable Mo. Duplicate digestions of O horizon and foliar samples agreed within 12 %, and duplicate digestions of bulk soil and rock samples agreed within 5 %.
Data analysis
Relationships among soil Mo pools, foliar and O horizon soil Mo concentrations, pH and other soil chemical parameters, state factors, and site characteristics were evaluated with least-squares linear regression. Differences in soil chemistry, site characteristics, and Mo concentrations between basaltic and sedimentary sites were determined using Student’s t-tests. All statistics were run in R version 3.1.0 (R Development Core Team 2014). Data normality was tested using the generic “qqnorm” function, which produces a normal quantile–quantile plot; non-normal data were transformed by adding one and taking the logarithm.
Results
Molybdenum concentrations and mobility in soil
Molybdenum concentrations ranged from 0.26 to 2.98 µg g−1 in basaltic bedrock and from 0.97 to 2.59 µg g−1 in sedimentary bedrock, and did not differ by bedrock lithology (Table 3). Bulk soil Mo concentrations were not correlated with underlying bedrock concentrations. In the top 10 cm, bulk soil Mo was slightly higher on sedimentary sites (p = 0.012), with a range of 0.69–1.97 µg g−1, and an average of 1.28 µg g−1, while basaltic sites had a range of 0.32–1.66 µg g−1 Mo with an average of 0.85 µg g−1 (Table 3).
There was no significant difference in extractable Mo pools based on bedrock type (Table 3; Fig. 2). Exchangeable and reducible Mo were the two smallest extractable pools. Overall, exchangeable Mo comprised just 1.4 ± 0.8 % (mean ± 1SD), and reducible Mo (RED 0.1 M) similarly comprised 1.4 ± 1.1 % of the total soil Mo. Oxidizable Mo (SOM2) was the largest extractable soil Mo pool, comprising on average 33 ± 16 % of the total soil Mo. The non-sequential extraction of oxidizable Mo (SOM1) yielded slightly less Mo, with a mean of 25 ± 13 % of the total soil Mo (ESM of Table A.1). Carbon concentrations measured before and after the oxidizable pool extraction showed that this extraction removed 77–88 % of soil C. Non-extractable Mo represented the largest fraction of soil Mo, 64 ± 17 % of the total soil Mo. Across all sites, pH was negatively correlated with several measures of soil Mo [exchangeable Mo: r2 = 0.40, p < 0.001, oxidizable Mo (SOM2): r2 = 0.40, p < 0.001, and bulk soil Mo: r2 = 0.13, p = 0.079 (only marginally significant)] (Fig. 3). In turn, pH was closely related to both base saturation and Al saturation, and the C and N content of the soil (Table 2). In contrast, there was no correlation between pH and reducible soil Mo.

Mean concentration of Mo from 0 to 10 cm depth increment in 11 basaltic and 11 sedimentary sites in the Oregon Coast Range in 3 selective chemical extractions, and the remaining resistant fraction (calculated by difference). Error bars represent 2 s.e. Results are not significantly different based on bedrock type for any of the 4 fractions

Extractable exchangeable Mo in 10’s of ng g−1 and oxidizable Mo in 100’s of ng g−1 (a) and bulk soil Mo in µg g−1 (b) versus soil pH (0–10 cm depth). Exchangeable regression in (a) shown in solid line: r2 = 0.40, p < 0.001, Oxidizable regression in (a) shown in dashed line: r2 = 0.40, p < 0.001. Bulk soil regression in (b): (r2 = 0.13, p = 0.079, only marginally significant)
Net mobility of Mo across the 22 mono-lithologic sites did differ between bedrock types (p = 0.25), and was characterized by high variability, with many sites showing a net gain of Mo relative to underlying parent material (Table 3). Based on τMoNb in the top meter of soil, mobility ranged from −0.51 (loss) to +4.15 (gain), with an average of +0.37 ± 1.05 (mean ± 1SD). Similarly, on the basis of just the top 10 cm, τMoNb ranged from −0.68 to +2.93, with an average of +0.13 ± 0.90 (mean ± 1SD).
Reducible Fe, Mn, and Al
We measured reducible Fe, Mn, and Al as an indicator of differences in secondary mineral abundance in soils formed on the two bedrock types. Overall, basaltic 0–10 cm soils were more Fe and Mn rich and had higher reducible Fe, Mn, and Al content, with 9493 and 6407 µg g−1 Fe, in basaltic and sedimentary sites, respectively, 1423 and 1059 µg g−1 Mn, respectively, and 10,423 and 7550 µg g−1 Al, respectively (ESM of Table A.2). The RED 0.1 M extraction was only 15 % more effective than the RED 0.04 M extraction for reducing Fe. Across all sites the average percentages of Fe, Mn, and Al extracted from the bulk soil by the RED 0.1 M extraction were 13, 86, and 11 % respectively.
Molybdenum in foliage, O horizon soil, and precipitation
Foliar Mo concentrations ranged from 2 to 82 ng g−1 across all sites, with an average Mo concentration of 19 ng g−1 and no difference based on bedrock type (Table 3). If one outlier was removed, foliar Mo was not correlated with bulk soil Mo concentration (p = 0.37), and was also not correlated with any particular soil extraction, total soil Mo content to 1 m depth (ESM of Table A.3.), soil C or N content, or soil pH. O horizon Mo concentrations were on average 7 times higher than foliage, ranging from 19 to 147 ng g−1 across all sites, with an average Mo concentration of 50 ng g−1 and no difference based on bedrock type. O horizon Mo was not correlated with bulk or extractable soil Mo pools, or with foliar Mo concentrations.
Multiplying the foliar Mo content by an average annual litterfall flux of 2.47 Mg ha−1 (Perakis et al. 2013) yields an approximate Mo flux from foliage to O horizon soil of 0.006 to 0.203 g ha−1 yr−1. Rainwater Mo concentrations across our north–south transect (sites 143, 7, and GV1) ranged from 4.8 to 9.8 pg ml−1, and across the east–west transect ranged from 3.5 to 19.5 pg ml−1 (Table 4). Using model-derived estimates of mean annual precipitation for all sites and an overall mean rainwater concentration of 7.7 pg ml−1, we estimate precipitation Mo inputs that range from 0.14 to 0.30 g ha−1 (Table 5).
Discussion
Mo partitioning and bulk soil concentrations
Our analysis of 24 surface soils from Oregon Coast Range forests found that approximately 1/3 of total Mo was susceptible to operationally defined selective metal extraction. These extractable pools are expected to be more rapidly cycled and bioavailable than the average of 2/3 of total Mo that was not extractable. Surprisingly, we found that little extractable Mo was associated with short-range-order Fe, Mn, and Al oxides (reducible), in contrast to prior studies focused on Fe oxy(hydr)oxide Mo adsorption as controlling Mo storage and bioavailability in soils (e.g., Karimian and Cox 1979; Goldberg et al. 1996; Gustafsson 2003). Instead, the largest fraction of extractable Mo we measured was associated with soil organic matter, based on the oxidizable extraction (H2O2). On average, oxidizable Mo represented 1/3 of the bulk soil Mo content across the Oregon Coast Range soils we sampled. This result agrees with a recent report of a topsoil sample from a hardwood forest in the New Jersey Pine Barrens, where 25 % of Mo was associated with organic matter (Wichard et al. 2009). This also agrees with results from soils on Maui and Iceland where the oxidizable Mo fraction comprised, on average, 18 and 30 %, respectively, of total soil Mo, a much larger fraction than was associated with short-range ordered Fe-oxides (King et al. 2014; Siebert et al. 2015). Although freely exchangeable Mo was only a small proportion of total Mo, this exchangeable and presumably highly-labile fraction was strongly correlated with the oxidizable Mo fraction (r2 = 0.77, p < 0.001), which suggests than the most easily mobilized Mo fraction in these soils is controlled by organic matter. Broadly, our findings identify organically bound fractions of Mo as crucial for both total Mo storage and cycling of freely available Mo in forest soils.
Andic properties are common in our soils, based on their classification as Andic Humudepts, Alic Hapludands, and Typic Fulvudands. The high intrinsic capacity for aggregation and buffering of these andic soils could have reduced the effectiveness of the NH2OH·HCl extraction in accessing Mo associated with reducible short-range ordered Fe, Al, and Mn oxy(hydr)oxides (Chadwick et al. 2003). However the reducible Mo pool was consistently small (average 1.4 % of bulk soil Mo) across all 24 sites. In contrast, this extraction yielded, on average, 80 % of the bulk soil Mn, 13 % of bulk soil Fe, and 20 % of bulk soil Al. While it is unlikely that all Fe and Al in soil is pedogenic, if we scale up to 100 % of Fe content as an upper-bound, then we estimate a maximum potential for Fe-oxide associated Mo that is still much smaller than the measured oxidizable pool (average 9.8 % of bulk soil Mo). It is possible that the large organic-associated Mo fraction observed in these soils may still interact with Fe oxide minerals, based on evidence that dissolved organic matter can cause micro-aggregation of iron oxides, resulting in a decreased penetration of MoO4 2− into iron-oxide micro-pores (Lang and Kaupenjohann 2003). This interaction has the potential to slow the process of molybdate immobilization on Fe-oxides, particularly in soils where organic matter content is high, and may contribute to the relatively low size of the reducible Mo pool observed at our sites.
The chemistry and mineralogy of underlying lithology often determines landscape patterns in the heavy metal contents of soils (Palumbo et al. 2000, Navas and Machin 2002). However, we did not observe correlations between soil and bedrock Mo concentrations across our 24 sites (p = 0.38), nor did we observe clear bedrock-related differences in soil Mo concentrations or in specific extractable soil Mo phases (Fig. 2). Together, these results imply that bedrock does not exert a strong control on soil Mo at these sites. Soil Mo also failed to correlate with site properties that can drive chemical weathering, such as mean annual precipitation, slope, aspect, and elevation (all p > 0.1). Additionally, soil Mo was not correlated with known indices of weathering in Coast Range soils such as effective cation exchange capacity or Hurst’s Redness Rating (Lindeburg et al. 2013). These results are unexpected given the varying mineralogy and relative mineral dissolution rates between the two lithologies. In the absence of an apparent parent material weathering effect on soil Mo, we consider atmospheric inputs and retention as the remaining dominant pedogenic processes likely controlling variation in soil Mo content across our sites.
Soil pH has often been invoked to control Mo mobility and bioavailability in soil through strong adsorption on Fe oxides (Karimian and Cox 1979; Gupta 1997; Goldberg and Forster 1998; Xu et al. 2006; Goldberg et al. 2008). However, we found that reducible Mo was not correlated with pH, in contrast to our expectations. Overall, the data show a pattern of higher Mo concentrations at lower pH, both in the bulk soil, and in the extractable exchangeable and oxidizable phases (Fig. 3). For the soils studied here, pH is correlated positively with organic matter content (r2 = 0.41, p = 0.003). It is possible that pH constraints on available Mo from reducible fractions are instead limited to soils with lower organic matter content than Oregon Coast Range soils, where soil C is relatively high for forests worldwide (Perakis and Sinkhorn 2011).
Mobility of soil Mo
Patterns in the mobility of Mo in soil based on loss or gain relative to the underlying parent material (τMoNb) were generally similar to those observed for soil Mo concentrations. On both a 0–10 and 0–100 cm basis, τMoNb correlated positively with the carbon content of the soil (Fig. 4), reinforcing the idea that Mo accumulation in the profile through organic matter processes dominates over leaching losses due to weathering of soil minerals. In contrast, τMoNb was not correlated with weathering indices or drivers, nor with the reducible Fe, Mn, or Al content across the 24 sites. Additionally, τMoNb was relatively invariant with depth in the soil down to 1 m (Supplementary content Table A.4), likely a reflection of efficient vertical mixing in Oregon Coast range soils (Anderson et al. 2002). The landscape-level picture emerging from our data therefore suggests that organic matter complexation is the dominant control of overall Mo retention in forest soils. As carbon appears to be a key control on soil Mo mobility, soil organic matter decomposition dynamics could therefore potentially control subsequent Mo remobilization.

Elemental mass transfer of Mo (τMoNb) in (a) the 0–10 cm depth increment and (b) the 0–100 cm depth increment, versus soil C pool (Mg ha−1). Linear regression in a: r2 = 0.58, p < 0.001), and b: r2 = 0.43, p < 0.001)
Foliar and O horizon soil Mo
Bioavailability can be evaluated by comparing extractable pools with foliar concentrations (e.g., Abedin et al. 2012). For the 24 Oregon Coast Range sites, however, foliar Mo in Douglas-fir did not correlate with any soil extractable pools, nor with bedrock Mo content, nor with either bulk mineral soil Mo from 0 to 10 cm, or total profile bulk soil Mo down to 1 m, on either a concentration or mass per unit area basis (Tables A.3, A.4). Other studies have similarly been unable to predict a bioavailable pool of Mo from either bulk soil concentrations (Natali et al. 2009), or other soil extraction schemes such as EDTA or hot pressurized H2O (Duvall et al. 2015), ion exchange resin extractable Mo (Jean et al. 2013), or oxalate extractable Mo (Karimaian and Cox 1979; Lang and Kaupenjohann 2000). These results, together with the data here, suggest that either (1) the specific soil chemical extractions used are not effectively targeting bioavailable phases, particularly for non-fixing plant species, or (2) that plant physiological regulation of Mo uptake and partitioning obscures any clear plant-soil Mo relationships.
As an alternative to foliar Mo reflecting soil bioavailability, foliar Mo may directly reflect plant demands, especially under high NO3 − conditions found at N enriched sites in the Oregon Coast Range (Perakis and Sinkhorn 2011). Douglas-fir can metabolize both NO3 − and NH4 +, although the availability of each ion in soil porewater can affect their relative uptake (Kammingavanwijk and Prins 1993). Because the reduction of NO3 − is the rate limiting step in its biological assimilation, and Mo is essential to the nitrate reductase enzyme, high soil NO3 − conditions can increase plant Mo requirements (Randall 1969; Kaiser et al. 2005). However, no relationship between soil N concentrations (which correlate closely with NO3 − availability, Perakis and Sinkhorn 2011) and foliar Mo was evident in our data.
Additionally, it remains unclear whether extractable Mo pools correlate with availability to soil microorganism; particularly N-fixing bacteria that exhibit Mo limitation in the Coast Range and elsewhere (Silvester 1989; Barron et al. 2008; Wurzburger et al. 2012; Reed et al. 2013). The recent discovery that the N-fixing bacteria Azotobacter spp. can use molybdophores to scavenge Mo from silicate minerals and Mo-tannic acid complexes calls historical definitions of bioavailability into question (Liermann et al. 2005; Wichard et al. 2009; Bellenger et al. 2011). Molybdophores contain the same catechol functional groups as soil organic matter, however the effectiveness of a molybdophore at weakening or breaking the bonds between soil organic matter and Mo may depend on the respective affinities of the two compounds for the Mo oxyanion. Thus, bioavailability of organically bound Mo is most likely a function of the size, structure, and solubility of the organic molecule it is bound to, which varies by soil organic matter source and stage of decomposition. An organically-bound Mo pool (as defined by the H2O2 extraction), which consists of both available and unavailable complexes, could help explain why Mo limitation has been observed Coast Range forest soils (Silvester 1989), even in the presence of large extractable Mo reservoirs. Additionally, heterotrophic bacteria are likely susceptible to small scale Mo depletion in local microenvironments (Maynard et al. 1994). Although organically-bound Mo may represent the largest pool of bioavailable Mo in soil, more research will be required to determine how much of the oxidizable Mo pool can be accessed by N-fixing organisms.
There are several possible reasons why we observed Mo concentrations that were 7 times higher in the O horizon soil than in Douglas-fir needles, a result similar to what has previously been observed in a mixed broadleaf-conifer forest in Pennsylvania (Kraepiel et al. 2015), and across conifer forests in Sweden (Brun et al. 2008). First, we standardized our sampling to the 1st year age cohort of needles, yet many elements increase in concentration as needles age (Tyler 2005). In Spruce trees, foliar Mo concentrations were found to range from 5 to 48 ng g−1 in first year needles, while needles from a 12-year- old spruce contained ~150 ng g−1 Mo (Lang and Kaupenjohann 2000). Second, an apparent enrichment of trace metals may occur prior to litterfall as the tree withdraws carbon and nutrients from the needles. Third, there may be an additional Mo source to the O horizon soil that increases Mo concentrations relative to litterfall from Douglas-fir. Vertical mixing of mineral soil into the O horizon is one potential source, but the extent of this mixing is expected to be highly variable across field sites. Furthermore, this mixing should dilute O-horizon C concentration and result in a negative correlation between Mo and C. The complete lack of correlation between O horizon soil C and Mo content (p = 0.96) suggests this mechanism is not likely to account for uniformly high Mo concentrations in O horizon soil relative to foliage.
Our estimates of Mo input from precipitation are, on average, 10 times larger than estimated Mo fluxes in litterfall (Table 5), raising the possibility that Mo enrichment in O horizon soil can also occur from adsorption of Mo from atmospheric deposition. Considering this over pedogenic timescales, Mo inputs from rainwater could be a substantial part of the overall Mo cycle, potentially contributing over 2000 g ha−1 of Mo per 10,000 years to Coast Range soil. Across our 24 sites, bulk mineral soil (0–10 cm) Mo pools averaged 533 g ha−1, with the highest concentrations being close to 1000 g ha−1. We did not quantify O horizon soil mass at these sites, but it varies little among similarly aged Douglas-fir forests in the Oregon Coast Range (mean = 12.87 Mg ha−1, s.e. = 0.43 Mg ha−1, n = 9, Perakis and Sinkhorn 2011). Using this value and our measured O horizon soil Mo concentrations, we estimate the O horizon pool of Mo averages 0.57 g of Mo ha−1, which could easily be influenced by an average rainfall Mo flux of 0.19 g ha−1 yr−1. Although atmospherically derived Mo would be susceptible to leaching or erosional loss, in organic rich soils where Mo adsorption and retention is high, this source is likely to be a dominant mechanism of Mo enrichment in soil relative to bedrock over thousands of years of pedogenesis.
Conclusions
Across temperate conifer forests of the Oregon Coast Range, the micronutrient Mo is retained in the soil profile primarily through adsorption to organic matter. Organically bound Mo is over 40 times larger than Mo bound to Fe and Mn oxides and accounts for an average of one-third of the bulk soil Mo concentrations. Both bulk soil Mo and extractable Mo fractions increase at low pH, concurrent with an increase in soil organic matter. Foliar Mo concentrations did not correlate to any soil extractable Mo pools or bulk soil Mo concentrations, suggesting that these chemical extractions do not represent available Mo for Douglas-fir foliage, or that foliar Mo concentrations are actively regulated by Douglas-fir. Atmospheric sources of Mo may be more important in subsidizing N2 fixation and other Mo-dependent biogeochemical processes than previously recognized, with input fluxes that greatly exceed internal recycling in the plant-soil system of forests. Our results raise the possibility that organic matter retention of atmospherically deposited Mo may shape patterns of Mo limitation of heterotrophic N2 fixation observed in surface organic soils of this region (Silvester 1989), but the fate of atmospherically derived Mo in soil and the degree to which it contributes to N2 fixation through time are yet to be determined.
Terrestrial N cycles are expected to respond strongly to rising atmospheric carbon dioxide, which stimulates plant productivity and may increase plant demand for bioavailable N (Hungate et al. 2003; van Groenigen et al. 2006, Wieder et al. 2015). However, as soils accumulate N (through biological fixation or anthropogenic deposition) accelerated N-cycling may compound the need for bioavailable Mo. These conditions could potentially increase biological Mo demand from soil. Understanding interactions between these macro- and micro- nutrients can improve predictions of how forests will respond to global change.
References
Abedin J, Beckett P, Spiers G (2012) An Evaluation of extractants for assessment of metal phytoavailability to guide reclamation practices in acidic soilscapes in northern regions. Can J Soil Sci 92(1):253–268
Anderson SP, Dietrich WE, Brimhall GH (2002) Weathering profiles, mass-balance analysis, and rates of solute loss: linkages between weathering and erosion in a small, steep catchment. GSA Bull 114:1143–1158
Barron AR et al (2008) Molybdenum limitation of asymbiotic nitrogen fixation in tropical forest soils. Nat Geosci 2(1):42–45
Bellenger JP, Wichard T, Xu Y, Kraepiel AM (2011) Essential metals for nitrogen fixation in a free-living N(2)-fixing bacterium: chelation, homeostasis and high use efficiency. Environ Microbiol 13(6):1395–1411
Bhella HS, Dawson MD (1972) The use of anion exchange resin for determining availible soil molybdenum. Soil Sci Soc Am 36:177–179
Bibak A, Borggard OK (1994) Molybdenum adsorption by aluminum and iron oxides and humic-acid. Soil Sci 158(5):323–328
Brimhall GH, Dietrich WE (1987) Constitutive mass balance relationship between chemical composition, volume, density, porosity, and strain in metasomatic hydrochemical systems—results On weathering and pedogenesis. Geochim Cosmochim Acta 51(3):567–587
Brun CB, Astrom ME, Peltola P, Johansson MJ (2008) Trends in major and trace elements in decomposing needle litters during a long-term experiment in Swedish forests. Plant Soil 306:199–210
Burt R (2004). Soil Survey Laboratory Methods Manual, Soil Survey Laboratory Investigations Report. National Soil Survey Center, Natural Resources Conservation Service U.S. Department of Agriculture Lincoln, Nebraska
Chadwick OA, Brimhall GH, Hendricks DM (1990) From a black box to a gray box a mass balance interpretation of pedogenesis. Geomorphology 3:369–390
Chadwick OA, Gavenda RT, Kelly EF, Ziegler K, Olson CG, Elliott WC, Hendricks DM (2003) The impact of climate on the biogeochemical functioning of volcanic soils. Chem Geol 202:195–223
Cox FR, Kamprath EJ (1972) Micronutrient soil tests. In: Mortvedt JJ, Giordano PM, Lindsay WL (eds) Micronutrients in agriculture. Soil Science Society of America, Madison, pp 289–318
Duvall BD, Natali SM, Hungate BA (2015) What constitutes plant-available molybdenum in sandy acidic soils? Commun Soil Sci PLant Anal 46(3):318–326
Goldberg S, Forster HS (1998) Factors affecting molybdenum adsorption by soils and minerals. Soil Sci 163(2):109–114
Goldberg S, Forster HS, Godfrey CL (1996) Molybdenum adsorption on oxides, clay minerals, and soils. Soil Sci Soc Am J 60(2):425–432
Goldberg S, Lesch SM, Suarez DL (2002) Predicting molybdenum adsorption by soils using soil chemical parameters in the constant capacitance model. Soil Sci Soc Am J 66:1836–1842
Goldberg S, Scalera E, Adamo P (2008) Molybdenum adsorption by volcanic Italian soils. Commun Soil Sci Plant Anal 39(5–6):693–706
Gupta UC (1997) Molybdenum in agriculture. Cambridge University Press, New York
Gustafsson JP (2003) Modelling molybdate and tungstate adsorption to ferrihydrite. Chem Geol 200:105–115
Heller PL, Peterman ZE, O’Neil JR, Shafiqullah M (1985) Isotopic provenance of sandstones from the Eocene tyee formation, oregon coast range. Geol Soc Am Bull 96:770–780
Hungate BA, Dukes JS, Shaw MR, Luo YQ, Field CB (2003) Nitrogen and climate change. Science 302(5650):1512–1513
Hurst VJ (1977) Visual estimation of iron in saprolite. Geol Soc Am Bull 88:174–176
Jarrell WM, Dawson MD (1978) Sorption and availability of molybdenum in soils of Western Oregon. Soil Sci Soc Am J 42(3):412–415
Jean ME, Phayvong K, Forest-Drolet J, Bellenger JP (2013) Molybdenum and phosphorus limitation of asymbiotic nitrogen fixation in forests of Eastern Canada: influence of vegetative cover and seasonal variability. Soil Biol Biochem 67:140–146
Kabata-Pendias A, Pendias H (2001) Trace elements in soils and plants, 3rd edn. CRC Press, Boca Raton
Kaiser BN, Gridley KL, Brady JN, Phillips T, Tyerman SD (2005) The role of molybdenum in agricultural plant production. Ann Bot 96(5):745–754
Kammingavanwijk C, Prins HBA (1993) The kinetics of NH4 + and NO3 − uptake by Douglas-fir from Single N-solutions and from solutions containing both NH4 + and NO3 −. Plant Soil 151(1):91–96
Karimian N, Cox F (1978) Adsorption and Extractability of molybdenum in relation to some chemical properties of soil. Soil Sci Soc Am Proceedings. 42:757–760
Karimian N, Cox F (1979) Molybdenum availability as predicted from selected soil chemical properties. Agron J 71(1):63–65
King EK, Thompson A, Hidges C, Pett-Ridge JC (2014) Towards understanding temporal and spatial patterns of molybdenum in the critical zone. Procedia Earth Planet Sci 10:56–62
Kraepiel AML, Dere AL, Herndon EM, Brantley SL (2015) Natural and anthropogenic processes contributing to metal enrichment in surface soils of central Pennsylvania. Biogeochemistry 123(1–2):265–283
Kubota J, Cary EE (1982) Cobalt, molybdenum, and selnium. In: Page AL (ed) Methods of soil analysis, Part 2, 2nd Ed. Monograph No 9. American Society of Agronomy, Madison, pp 485–500
Kurtz AC, Derry LA, Chadwick OA, Alfano MJ (2000) Refractory element mobility in volcanic soils. Geology 28:683–686
Lang F, Kaupenjohann M (1998) Influence of liming and kieserite fertilization on the molybdenum dynamics of forest sites. Forstwiss Cent 17(6):316–326
Lang F, Kaupenjohann M (2000) Molybdenum at German Norway spruce sites: contents and mobility. Can J For Research-Revue Can De Rech For 30(7):1034–1040
Lang F, Kaupenjohann M (2003) Immobilization of molybdate by iron oxides: effects of organic coatings. Geoderma 113(1–2):31–46
Liermann LJ, Guynn RL, Anbar A, Brantley SL (2005) Production of a molybdophore during metal-targeted dissolution of silicates by soil bacteria. Chem Geol 220(3–4):285–302
Lindeburg KS, Almond P, Roering JJ, Chadwick OA (2013) Pathways of soil genesis in the Coast Range of Oregon, USA. Plant Soil 367:57–75
Lombin G (1985) Micronutrient soil tests for semi-arid savanna of Nigeria: boron and molybdenum. Soil Sci Plant Nutr 34(1):1–11
Marks JA, Pett-Ridge JC, Perakis SS, Allen JL, McCune B. (in press) Response of the nitrogen-fixing lichen Lobaria pulmonaria to phosphorus, molybdenum, and vanadium. Ecosphere
Maynard RH, Premakumar R, Bishop PE (1994) Mo-independent nitrogenase 3 is advantageous for diazotrophic growth of Azotobacter-vinlandii on solid medium containing molybdenum. J Bacteriol 176:5583–5586
McBride MB, Richards BK, Steenhuis T, Spiers G (2000) Molybdenum uptake by forage crops grown on sewage sludge-amended soils in the field and greenhouse. J Environ Qual 29(3):848–854
McWilliams RG (1973) Stratigraphic and bio-stratigraphic relationships of the Tyee and Yamhill formations in central western Oregon. Oregon Dep Geol Miner Ind, Ore Bin 35(11):169–186
Menge DNL (2011) Conditions under which nitrogen can limit steady-state net primary production in a general class of ecosystem models. Ecosystems 14(4):519–532
Mortvedt JJ, Anderson OE (1982) Forage legumes: Diagnosis and correction of molybdenum and manganese problems. Southern Cooperative Series Bulletin, vol 178. University of Georgia, Athens
Natali SM, Sanudo-Wilhelmy SA, Lerdau MT (2009) Effects of elevated carbon dioxide and nitrogen fertilization on nitrate reductase activity in sweetgum and loblolly pine trees in two temperate forests. Plant Soil 314(1–2):197–210
Navas A, Machin J (2002) Spatial distribution of heavy metals and arsenic in soils of Aragon (northeast Spain): controlling factors and environmental implications. Appl Geochem 17(8):961–973
Neunhauser C, Berreck M, Insam H (2001) Remediation of soils contaminated with molybdenum using soil amendments and phytoremediation. Water Air Soil Pollut 128:85–96
NRCS (2009) Web soil survey. http://www.websoilsurvey.ncsc.usda.gov/app/. Accessed 29 Oct 2009
Palumbo B, Angelone M, Bellanca A, Dazzi C, Hauser S, Neri R, Wilson J (2000) Influence of inheritance and pedogenesis on heavy metal distribution in soils of Sicily, Italy. Geoderma 95(3–4):247–266
Perakis SS, Sinkhorn ER (2011) Biogeochemistry of a temperate forest nitrogen gradient. Ecology 92:1481–1491
Perakis S et al (2006) Coupled nitrogen and calcium cycles in forests of the Oregon Coast Range. Ecosystems 9(1):63–74
Perakis SS, Matkins JJ, Hibbs DE (2012) N-2-fixing red alder indirectly accelerates ecosystem nitrogen cycling. Ecosystems 15(7):1182–1193
Perakis SS et al (2013) Forest calcium depletion and biotic retention along a soil nitrogen gradient. Ecol Appl 23(8):1947–1961
R Development Core Team (2014) R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria
Randall PJ (1969) Changes in nitrate and nitrate reductase levels on restoration of molybdenum to molybdenum-deficient plants. Aust J Agric Res 20(4):635–642
Rauret G et al (1999) Improvement of the BCR three-step sequential extraction procedure prior to the certification of new sediment and soil reference materials. J Environ Monit 1(1):57–61
Reed SC, Cleveland CC, Townsend AR (2013) Relationships among phosphorus, molybdenum and free-living nitrogen fixation in tropical rain forests: results from observational and experimental analyses. Biogeochemistry 114(1–3):135–147
Siebert C, Pett-Ridge JC, Opfergelt S, Guicharnaud RA, Halliday AN, Burton KW (2015). Molybdenum isotope fractionation in soils: Influence of redox conditions, organic matter, and atmospheric inputs. Geochemica et Cosmochimica Acta. (in press)
Silvester WB (1989) Molybdenum limitation of asymbiotic nitrogen fixation in forests of Pacific Northwest America. Soil Biol Biochem 21(2):283–289
Snavely P Jr, MacLeod N (1974) Yachats Basalt—An upper eocene differentiated volcanic sequence in the Oregon Coast Range. J Res US Geol Surv 2(4):395–403
Snavely PD Jr, Wagner HC, MacLeod NS (1964) Rhythmic-bedded Eugeosynclinal deposits of the Tyee formation, Oregon Coast Range. Kans Geol Surv Bull 169:461–480
Snavely PD, MacLeod NS, Wagner HC (1968) Tholeiitic and alkalic basalts of the Eocene Siletz River Volcanics. Oregon Coast Range. Am J Sci 266:454–481
Spies TA et al (2002) The ecological basis of forest ecosystem management in the Oregon Coast Range. In: Hobbs SD, Hayes JP, Johnson RL, Reeves GH, Spies TA, Tappeiner JC II, Wells GE (eds) Forest and stream management in the Oregon Coast Range. Oregon State University Press, Corvallis, pp 31–67
Stiefel EI (2002) The biogeochemistry of molybdenum and tungsten. Met Ions Biol Syst 39:1–29
Tessier A, Campbell PGC, Bisson M (1979) Sequential extraction procedure for the speciation of particulate trace metals. Anal Chem 51(7):844–851
Thomas RQ, Canham CD, Weathers KC, Goodale CL (2010) Increased tree carbon storage in response to nitrogen deposition in the US. Nat Geosci 3(1):13–17
Tyler G (2005) Changes in the concentrations of major, minor and rare-earth elements during leaf senescence and decomposition in a Fagus sylvatica forest. For Ecol Manage 206(1–3):167–177
Ure AM, Quevauviller P, Muntau H, Griepink B (1993) Speciation of heavy metals in soils and sediments- an account of the improvement and harmonization of extraction Techniques undertaken under the auspices of the BCR of the commission of the European Communities. Int J Environ Anal Chem 51(1–4):135–151
van Groenigen KJ et al (2006) Element interactions limit soil carbon storage. Proc Natl Acad Sci USA 103(17):6571–6574
Vitousek PM, Howarth RW (1991) Nitrogen limitation on land and in the sea how can it occur. Biogeochemistry 13(2):87–115
Wang T, Hamann A, Spittlehouse D (2009) ClimateWNA. University of British Columbia, Department of Forest Sciences, Centre for Forest Conservation Genetics, Vancouver, BC. http://www.genetics.forestry.ubc.ca/cfcg/ClimateWNA_web/. Accessed May 2012
Wichard T, Mishra B, Myneni SCB, Bellenger JP, Kraepiel AML (2009) Storage and bioavailability of molybdenum in soils increased by organic matter complexation. Nat Geosci 2(9):625–629
Wieder WR, Cleveland CC, Smith WK, Todd-Brown K (2015) Nutrient availability strongly constrains future terrestrial productivity and carbon storage. Nat Geosci. doi:10.1038/ngeo2413
Wurzburger N, Bellenger JP, Kraepiel AM, Hedin LO (2012) Molybdenum and phosphorus interact to constrain asymbiotic nitrogen fixation in tropical forests. PLoS ONE 7(3):e33710
Xu N, Christodoulatos C, Braida W (2006) Adsorption Of molybdate and tetrathiomolybdate onto pyrite and goethite: effect of pH and competitive anions. Chemosphere 62(10):1726–1735
Acknowledgments
We gratefully acknowledge Justin Hynicka for field site selection, collecting the soil, bedrock, and foliar samples, and for sample analysis on bedrock and bulk soil. Andy Ungerer of the Oregon State University Keck Collaboratory for Plasma Spectrometry, and Chris Catricala, Lauren Armony, Clarinda Wilson, and George Pope are thanked for assistance with lab analyses. We thank Kurt Smemo and two anonymous reviewers for comments which improved the manuscript. Any use of trade names is for descriptive purposes only and does not imply endorsement by the U.S. Government. NSF award 1053470 to Pett-Ridge supported this work.
Author information
Authors and Affiliations
Corresponding author
Additional information
Responsible Editor: Melany Fisk.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Marks, J.A., Perakis, S.S., King, E.K. et al. Soil organic matter regulates molybdenum storage and mobility in forests. Biogeochemistry 125, 167–183 (2015). https://doi.org/10.1007/s10533-015-0121-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10533-015-0121-4