
Abstract
The dechlorinating Dehalococcoides mccartyi species requires acetate as carbon source, but little is known on its growth under acetate limiting conditions. In this study, we observed growth and dechlorination of a D. mccartyi-containing mixed consortium in a fixed-carbon-free medium with trichloroethene in the aqueous phase and H2/CO2 in the headspace. Around 4 mM formate was produced by day 40, while acetate was constantly below 0.05 mM. Microbial community analysis of the consortium revealed dominance by D. mccartyi and Desulfovibrio sp. (57 and 22% 16S rRNA gene copies, respectively). From this consortium, Desulfovibrio sp. strain F1 was isolated and found to produce formate and acetate (1.2 mM and 48 µM, respectively, by day 24) when cultivated alone in the above mentioned medium without trichloroethene. An established co-culture of strain F1 and D. mccartyi strain 195 demonstrated that strain 195 could grow and dechlorinate using acetate produced by strain F1; and that acetate was constantly below 25 µM in the co-culture. To verify that such low level of acetate is utilizable by D. mccartyi, we cultivated strain 195 alone under acetate-limiting conditions and found that strain 195 consumed acetate to below detection (5 µM). Based on the acetate consumption and cell yield of D. mccartyi, we estimated that on average 1.2 × 108 acetate molecules are needed to supply carbon for one D. mccartyi cell. Our study suggests that Desulfovibrio may supply a steady but low amount of fixed carbon to dechlorinating bacteria, exhibiting important implications for natural bio-attenuation when fixed carbon is limited.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Chlorinated ethenes including tetrachloroethene (PCE), trichloroethene (TCE), and their conversion products dichloroethene (DCE) and vinyl chloride (VC) are among the most common groundwater contaminants (Abelson 1990). Substantial laboratory and field studies have been performed on biological reductive dechlorination of chloroethenes and on identifying/characterizing organohalide respiring bacteria. So far, a variety of anaerobic genera, including Desulfuromonas, Dehalobacter, Desulfitobacterium, Enterobacter, and Sulfurospirillum, are known to be capable of dechlorinating PCE to DCEs in the presence of electron donors/fixed carbon sources such as lactate, pyruvate, formate, and acetate (Finneran et al. 2002; Holliger et al. 1998; Krumholz et al. 1996; Scholz-Muramatsu et al. 1995; Sharma and McCarty 1996). Reductive dechlorination of chloroethenes has rarely been investigated under autotrophic conditions. It has been reported that Desulfuromonas sp. strain BB1 can dechlorinate PCE to cis-DCE via consumption of acetate generated from H2/CO2 by an acetogen—Sporomusa ovata (He et al. 2002).
Further dechlorination of DCEs to ethene has only been described for one genus of bacteria—Dehalococcoides, whose genomic sequences have revealed obligate requirements for acetate as carbon source, H2 as electron donor, and halogenated compounds as electron acceptors (Löffler et al. 2013). The type strain, Dehalococcoides mccartyi strain 195, was reported to grow in completely defined mineral salts medium amended with acetate, H2/CO2 and TCE (He et al. 2007), and was incapable of dechlorinating TCE in the absence of either acetate or H2. To date, the reductive dechlorination of PCE and/or TCE past cis-DCE to VC and ethene has not been investigated in an autotrophic consortium via syntrophy. However, in practice, in situ bioremediation is commonly performed by microbial consortia containing dechlorinators and other beneficial microorganisms, and natural bio-attenuation would require autotrophic activity because many chloroethene-contaminated subsurface environments are naturally oligotrophic (Schneidewind et al. 2014; Ramsburg et al. 2004).
When D. mccartyi is grown in enriched cultures or established consortia, it commonly utilizes products (e.g., acetate and H2) generated by fermenting organisms such as Desulfovibrio, Acetobacterium, and Clostridium (He et al. 2007; Ritalahti and Löffler 2004; Richardson et al. 2002; Aulenta et al. 2005; Men et al. 2012). In particular, sulfate-reducing bacteria Desulfovibrio are typically found to co-exist with D. mccartyi and are metabolically versatile. For example, Desulfovibrio desulfuricans can utilize both lactate and pyruvate for energy generation via fermentative pathways in the absence of exogenous terminal electron acceptors, producing acetate, CO2, and H2, while Desulfovibrio vulgaris strain Hildenborough can transform lactate to acetate and H2 when grown syntrophically with D. mccartyi or a H2-utilizing methanogen (Men et al. 2012; McInerney et al. 1981; Schink 1997).
There are also reports of autotrophic Desulfovibrio growth, with at least three confirmed strains. Desulfovibrio sp. strain HRM1 (Brysch et al. 1987) and Desulfovibrio indonesiensis strain P23 (Fichtel et al. 2012) grow with H2/CO2 in the presence of sulfate. However, the volatile fatty acid (VFA) products resulting from their autotrophic growth were not identified. The third strain Desulfovibrio sp. strain G11 was claimed to be an autotroph, but no supporting experimental data were presented (McInerney et al. 1981; Dolfing et al. 2008). Mixotrophic growth with H2/CO2 and acetate is more common with Desulfovibrio bacteria, e.g., D. desulfuricans (Londry and Marais 2003; Mechalas and Rittenberg 1960) and D. vulgaris (Peters et al. 1999). Notably, formate was formed as an intermediate when D. vulgaris was grown chemolithoheterotrophically with H2/CO2 and acetate under sulfate-limiting conditions (Peters et al. 1999). To date, autotrophic sulfate reducers fall into two categories by the mechanisms they utilize to synthesize acetyl-CoA from CO2—the reductive citric acid cycle and the reductive CO-dehydrogenase pathway (acetyl-CoA pathway) (Rabus et al. 2006). However, no information is available on autotrophic growth and its VFA products by Desulfovibrio autotrophs. The carbon fixing mechanisms that they utilize also remain elusive.
In this study, we characterized an autotrophic dechlorinating consortium (culture EC195) from which D. mccartyi strain 195 was isolated. We aimed to determine the actual carbon source for growth of D. mccartyi in culture EC195. Acetate utilization patterns by D. mccartyi including acetate utilization threshold and cell yield per mole acetate were also investigated.
Materials and methods
Chemicals
Analytical grade chloroethenes including TCE, DCEs, and VC were purchased from Sigma-Aldrich-Fluka (St. Louis, MO) or Supelco (Bellefonte, PA). Ethene was obtained from Alltech Associates, Inc. (Deerfield, IL). Other chemicals used in this study were obtained from Sigma-Aldrich-Fluka (St. Louis, MO). Gases (air, nitrogen, H2, and CO2) were supplied by Praxair, Inc. (Berkeley, CA) and Soxal (Singapore).
Culture growth conditions and cultivation history of culture EC195
Concentrations of volatile compounds such as TCE in this study are presented as nominal concentrations, i.e., total molar amounts in aqueous and gaseous phases divided by the aqueous phase volume. All cultures were prepared in 160 mL serum bottles with 100 mL bicarbonate-buffered medium (containing 30 mM sodium bicarbonate) and 60 mL headspace. All cultures received H2 (20 mL, or 0.33 atm) in the headspace as electron donor on top of the original N2/CO2 headspace (final headspace pressure 1.33 atm), unless specified otherwise. All experimental bottles in this study were incubated in the dark at 30 °C without shaking.
Culture EC195 is a subculture of the parent culture from which D. mccartyi strain 195 was isolated (Maymó-Gatell et al. 1997). It was initially cultivated in an anaerobic medium containing activated sludge extract of undefined composition (Maymó-Gatell et al. 1997), before being transferred (2% [v/v]) to defined mineral salts medium spiked with TCE (~ 0.5 mM) as electron acceptor and acetate as carbon source as described previously (He et al. 2003). Acetate was then omitted from the medium recipe for the next 15 transfers while other growth conditions remained the same. The still-actively-dechlorinating culture EC195 was then transferred for another five times in a modified medium with 50 times higher amount of vitamin B12 (final concentration 50 µg L−1) (He et al. 2007) and then was inoculated into triplicate bottles (3% [v/v]) for dechlorination kinetics studies. The recipe of the final basal medium without TCE or additional fixed carbon amendments was referred to as DCB-1 medium. Due to oxidation of added reductants—l-cysteine, dithiothreitol (DTT), and Na2S, the DCB-1 medium may contain a trace amount of sulfate (< 0.05 mM detection limit, see below) or other sulfur-containing compounds before inoculation (Chen and Morris 1972).
Pure culture of D. mccartyi strain 195 was kindly provided by Dr. Stephen H. Zinder of Cornell University (Ithaca, NY) and was used for all experiments that involved strain 195 in this study. Desulfovibrio sp. strain F1 was isolated from culture EC195 and was grown in DCB-1 medium amended with sulfate (2 mM) and H2 (20 mL, or 0.33 atm) in the headspace, resulting in a H2/sulfate molar ratio of 4.9, unless specified otherwise.
To establish an autotrophic co-culture, active inocula of Desulfovibrio sp. strain F1 (1% [v/v]) and D. mccartyi strain 195 (2% [v/v]) were transferred to DCB-1 medium amended with ~ 0.5 mM TCE. Before inoculation, both inocula were rinsed three times under anaerobic conditions with sterilized DCB-1 medium by centrifugation at 10,000×g for 15 min and removing the supernatant, in order to minimize the organic compounds carried over into the cultures.
Analytical techniques
Chloroethenes and ethene were measured with an Agilent gas chromatograph (GC7890) equipped with a flame ionization detector (GC-FID). Headspace samples (100 µL) that were withdrawn by using a gas-tight glass syringe (model 1725; Hamilton Co., Reno, NV) were separated on a GS-GasPro column (30 m by 0.32 mm; J&W Scientific) as previously described (Ding et al. 2013). Volatile fatty acids (VFAs) including formate and acetate were determined on an Agilent 1260 HPLC system (Agilent, CA, USA) with a UV detector (210 nm). Separation of VFAs was conducted on a Rezex ROA-Organic Acid H+ (8%) HPLC column (Phenomenex) at room temperature, with 2.5 mM sulfuric acid as a mobile phase at 0.5 mL min−1 flow rate (10 or 100 µL injection volume). Fluorescence microscopy was performed on a Nikon Eclipse 200 using Syto-9 staining dye (Invitrogen). Scanning electron microscopy was performed as described previously (Ding et al. 2014). In short, cells were harvested by filtration through a 0.2 μm pore size polycarbonate membrane and fixed with 2.5% glutaraldehyde before viewing on a Philips XL 30 scanning electron microscope (FEI, Hillsboro, OR, USA). Sulfate in the culture medium was precipitated first with barium chloride and the resulting suspension was well mixed and measured for absorbance at 420 nm on a Tecan Infinite® 200 PRO microplate reader (Switzerland) (detection limit 0.05 mM). H2 in the headspace was detected using an Agilent GC7890 equipped with a thermal conductivity detector as described previously (detection limit 8 ppmv) (Bramono et al. 2011).
Molecular analyses
Genomic DNA was extracted from actively growing cultures by using an Ultra Clean Microbial DNA Kit (MO BIO Laboratories, Inc., Carlsbad, CA) according to the manufacturer’s recommendations. Quantitative real-time polymerase chain reaction (qPCR) on D. mccartyi and Desulfovibrio was performed on an ABI 7500 Fast real-time PCR System (ABI, Foster City, CA) by using QuantiTect SYBR green PCR master mix (QIAGEN, Germany) and primers Dhc-forward/reverse (5′-GGT AAT ACG TAG GGA AGC AAG CG-3′ and 5′-CCG GTT AAG CCG GGA AAT T-3′) (for D. mccartyi) (Holmes et al. 2006) and Dsv691F/826R (5′-CCG TAG ATA TCT GGA GGA ACA TCA G-3′ and 5′-ACA TCT AGC ATC CAT CGT TTA CAG C-3′) (for Desulfovibrio) (Fite et al. 2004). Quantification of total cells in the enrichment cultures by qPCR was performed by targeting the genomic DNA with universal primers 338F and 518R (5′-ACT CCT ACG GGA GGC AGC AG-3′ and 5′-ATT ACC GCG GCT GCT GG-3′) (Muyzer et al. 1993; Lane 1991).
Construction of 16S rRNA gene clone library, restriction enzyme digestion by HhaI and MspI (NEB, Ipswich, MA, USA), restriction fragment length polymorphism, and sequencing of representative plasmids were done as described previously (Cheng et al. 2010). The 16S rRNA gene sequences of Desulfovibrio sp. strain F1 were deposited in GenBank under the accession numbers KC120814 and KC120815.
PCR amplification and DGGE was performed by using primers 341FGC and 518R (5′-CGC CCG CCG CGC GCG GCG GGC GGG GCG GGG GCA CGG GGG GCC TAC GGG AGG CAG CAG-3′ and 5′-ATT ACC GCG GCT GCT GG-3′) (Muyzer et al. 1993) as described previously (Cheng et al. 2010).
Results
Chloroethene dechlorination by an autotrophic culture EC195
Culture EC195 exhibited autotrophic growth and stable TCE dechlorination activities after being transferred into the fixed-carbon-free DCB-1 medium amended with TCE. A clone library analysis indicated that D. mccartyi species (57%) and Desulfovibrio species (22%) are the two dominant populations in culture EC195 (Table 1). Spirochaeta also occurred in culture EC195, which has commonly been found in other dechlorinating enrichment cultures (Men et al. 2017; Kaufhold et al. 2013).
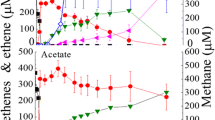
Dechlorination of TCE by culture EC195 started with a 15-day lag phase, after which ~ 0.5 mM TCE was dechlorinated to VC and ethene via cis-DCE as the intermediate within 35 days (Fig. 1). Formate production was observed in the EC195 culture, reaching ~ 4 mM after 40 days (Fig. 1a). Accompanying growth and dechlorination was the consumption of H2 which was indicated by decreasing headspace concentrations. The cell densities of D. mccartyi and total bacteria in culture EC195 increased ~ 27-fold and ~ 21-fold to a maximum of 2.9 × 107 and 1.08 × 108 cells mL−1, respectively (Fig. 1b).

Dechlorination of TCE by culture EC195 in DCB-1 medium without fixed carbon amendments. a Dechlorination of TCE and production of formate by culture EC195. b Growth profile of total cells versus strain 195 in culture EC195 during dechlorination of TCE to VC and ethene. Ethene (< 0.02 mM) was not shown. Error bars (± SD) were shown if not smaller than the symbols. All data points were averaged from triplicate bottles
It was interesting that acetate was not detected throughout the test (detection limit 0.05 mM), although it was reported to be indispensable for D. mccartyi species (Löffler et al. 2013). The possibility of formate acting as the carbon source for D. mccartyi seemed low, as D. mccartyi strain 195 did not grow or dechlorinate when 4 mM formate was added to the DCB-1 medium (data not shown), which was also consistent with the previous report (Maymó-Gatell et al. 1997). Moreover, when strain 195 was cultivated alone in DCB-1 medium with TCE but without fixed carbon, no growth or dechlorination activity was observed and no formate or other VFAs were detected, even after 3-months incubation (data not shown).
Isolation of an autotrophic Desulfovibrio sp. strain F1 from culture EC195
Based on the above observations, we hypothesized that the required fixed carbon for D. mccartyi and the formate produced were generated autotrophically by one of the dominant species—Desulfovibrio, although some low abundance organisms might also produce fixed carbon.
To enrich Desulfovibrio which was presumably a sulfate reducer, culture EC195 was transferred in DCB-1 medium amended with 10 mM acetate and 2 mM sulfate, but without TCE. This culture was later subjected to serial dilution. Three repeated serial dilutions until 10−8 resulted in a culture with only vibrio-shape cells under fluorescence microscope. After another three rounds of colony picking from agar shakes and immediate serial dilution, an isolate of Desulfovibrio was obtained, which was designated strain F1. Cells of strain F1 have a dimension of 1–1.5 µm length and 0.3–0.5 µm width (Fig. S1). Cells are motile with monotrichous flagella (Fig. S1B). A 16S rRNA gene clone library of 80 clones showed a single digestion pattern using either MspI or HhaI as the restriction enzymes. DGGE analysis on 16S rRNA genes exhibited two bands for strain F1, both of which were affiliated with Desulfovibrio after excision and sequencing (Lane 3, Fig. S2). Analysis of 22 clones from the above-mentioned clone library on DGGE gel showed that 15 of them belong to type 1 (top, thicker band, accession number KC120814) and the remaining 7 belong to type 2 (bottom, weaker band, accession number KC120815) (DGGE picture not shown). The closest reported relative of strain F1 is Desulfovibrio desulfuricans strain Ser-2 (identical base pairs: type 1—1397/1400, type 2—1399/1400).
Strain F1 was then transferred to DCB-1 medium with 2 mM sulfate but without acetate. Within 15 days of incubation, black precipitates appeared which are typical of metal sulfides, and the headspace pressure of the bottles dropped quickly, indicating the consumption of H2. After more than 5 transfers (3% [v/v] inocula) in the same medium, the cultures still showed the same activity and the autotrophic culture has been maintained for more than 3 years. The possibility that reductant l-cysteine and DTT may have served as the fixed carbon source for Desulfovibrio was excluded by observing similar cell growth and formate formation in the l-cysteine- and DTT-free medium where sodium sulfide (Na2S) served as the only reductant.
Cell growth of strain F1 requires H2 and sulfate, and was positively correlated to initial sulfate concentrations from 0 to 2 mM (Table 2), a typical characteristic of hydrogenotrophic sulfate reducer (4 H2 + SO42− + H+ → HS− + 4 H2O) (Muyzer and Stams 2008). In set “S0” where no sulfate was added, there was still moderate cell growth (0.17 × 108 16S rRNA gene copy mL−1) which corresponded to ~ 0.03–0.05 mM sulfate if we assume a linear correlation between sulfate concentrations and cell growth. This agreed with previous estimation that sulfate concentration was < 0.05 mM. The highest level of sulfate tested (6 mM for “S4” set in Table 2) caused the pH to reach ~ 8.0 by day 16 and inhibited cell growth, probably through production of sulfide. For experimental sets with initial sulfate of 0.2–2 mM, sulfate concentrations quickly dropped to below the detection limit (0.05 mM) within the first 8 days, while bottles with 6 mM sulfate still contained ~ 2 mM on day 8.
Formate production by strain F1 was also dependent on H2 and sulfate (Table 2). The highest formate concentration (9.83 ± 0.30 mM) was observed on day 24 in bottles supplied with 30 mL (0.5 atm) H2 and 0.6 mM sulfate. Acetate, which was thought to stimulate cell growth as described by Brysch et al. (1987), did not have any significant impact on cell growth or formate production, suggesting that acetate possibly does not serve as carbon source or electron donor for strain F1 under the tested conditions (set “AS−” and “AS+” in Table 2). Mass balances of the “H1” bottles [10 mL (0.16 atm) H2 and 2 mM sulfate] showed that on day 16, 2.8 ± 0.2 mL H2 was left of the initial 10 mL, while 1.66 ± 0.4 mM formate was formed and sulfate was reduced from 2–< 0.05 mM. The consumed 7.2 mL H2 (equivalent aqueous concentration 9.8 mM) is close to the theoretical H2 consumption of 9.7 mM (assuming that formate production requires one molecule of H2 and sulfate reduction requires four).
Desulfovibrio sp. strain F1 produces trace amounts of acetate
Although strain F1 has been confirmed to grow autotrophically, its relationship with D. mccartyi in culture EC195 remained unclear, because acetate was not detected in the culture liquid of strain F1 with the initial acetate detection protocol using HPLC–UV (detection limit: 0.05 mM). Acetate was again measured after decreasing the detection limit to 5 µM by increasing the HPLC injection volume from 10 to 100 µL. Strain F1 was then grown in DCB-1 medium without sulfate amendment and with 30 mL (0.5 atm) H2 (identical condition as “S0” set in Table 2). Using the revised acetate detection method, we did observe production of acetate which reached 48 ± 3 µM by day 42, together with a formate concentration of 1.2 ± 0.1 mM (Fig. 2).

Production of trace amounts of acetate by Desulfovibrio sp. strain F1 in DCB-1 medium without fixed carbon amendments. Cultures were grown in 30 mL medium with 40 mL headspace, and were inoculated from parent cultures that had been maintained in DCB-1 medium for at least five transfers. H2 (30 mL) was added as electron donor. All data points were averaged from triplicate bottles
Chloroethene dechlorination by an autotrophic co-culture of strain F1 and strain 195
A co-culture of strain 195 (2% [v/v]) and strain F1 (1% [v/v]) was established to verify whether strain F1 supplies the required carbon source (acetate) for the growth and dechlorination of strain 195 in the DCB-1 medium without fixed carbon amendment. This co-culture was transferred in DCB-1 medium amended with H2 and TCE at least five times, and constantly showed TCE dechlorination and VFA production activities.
In DCB-1 medium amended with TCE, the co-culture dechlorinated ~ 0.62 mM TCE to VC and ethene within 20 days (Fig. 3a), exhibiting a similar but faster dechlorination profile compared with culture EC195. On day 20, strain 195 and strain F1 reached cell densities of 4.5 × 108 and 2.3 × 107 16S rRNA gene copies mL−1, respectively, representing 126-fold and 39-fold increases compared to the cell densities upon inoculation (Fig. 3b). Cell densities of strain F1 reached its maximum on day 3 and remained stagnant through the remainder of the experiment. Accompanying the TCE dechlorination, formate accumulated to 1.2 mM in the DCB-1 medium while acetate peaked at day 3 (24 µM) and then slowly dropped to below 5 µM (Fig. 3c). Therefore, in this autotrophic co-culture, Desulfovibrio sp. strain F1 fixes the inorganic carbon to support the growth and dechlorination of strain 195 in the form of acetate.

Dechlorination of TCE by an established autotrophic co-culture in DCB-1 medium without fixed carbon amendments. a Dechlorination of TCE by the co-culture. b Growth profile of D. mccartyi and Desulfovibrio cells during dechlorination of TCE (Dhc: D. mccartyi strain 195, Dsv: Desulfovibrio sp. strain F1). c Production of acetate and formate during TCE dechlorination by the co-culture. Ethene (< 0.02 mM) was not shown. Error bars (± SD) were shown if not smaller than the symbols. All data points were averaged from triplicate bottles
Acetate utilization threshold by D. mccartyi strain 195
To verify that the low level acetate (< 50 µM) is indeed utilizable for D. mccartyi, we grew D. mccartyi strain 195 in various acetate concentrations and monitored acetate consumption during dechlorination of ~ 0.63 mM TCE (Fig. 4). When initial acetate concentrations were 157 ± 9 µM or higher (Fig. 4a, b), strain 195 consumed 93 ± 7 µM acetate, producing VC and ethene (0.148 mol of acetate uptake per mole of TCE dechlorinated), with final cell densities of 4.8 × 108 mL−1 (Fig. 4f). For initial acetate concentrations of 72 ± 2 µM or lower (Fig. 4c, d), the growth of strain 195 became limited and nearly all the acetate was consumed to a level below 5 µM. The extent of TCE dechlorination was also lower than those with higher acetate concentrations. No dechlorination activity was observed if no acetate was provided (Fig. 4e). In control bottles with 303 ± 7 µM acetate but without inocula, no change in acetate concentration was observed throughout the experimental period (data not shown). Initial acetate concentrations higher than 157 µM (up to 10 mM tested) had no enhanced effect on TCE dechlorination rates/extents (data not shown). Under acetate limiting conditions (72 and 45 µM), the final cell densities achieved were significantly lower than those when acetate was abundant (157 and 292 µM), and were positively correlated with the amount of acetate consumed (Fig. 4f). On day 8 when all acetate was consumed to below 5 µM, there was still TCE left that was further converted to VC under acetate limiting conditions (Fig. 4c, d), showing that dechlorination continued after cell growth ceased.

Comparison of D. mccartyi strain 195 grown with various concentrations of acetate. a–e Dechlorination kinetics and acetate utilization profiles of D. mccartyi strain 195. Initial acetate concentrations are 292 µM, 157 µM, 72 µM, 45 µM, and 0 µM, respectively for (a–e). Acetate concentrations in (e) never exceeded detection limit and therefore are not shown. Open circles—acetate; open squares—TCE; closed circles—DCEs; closed triangles—VC; closed squares—ethene. f Final cell density and acetate consumption by strain 195 at conditions described in (a–e). All data points are averages of triplicate bottles
Discussion
Desulfovibrio sp. strain F1 was isolated in this study, an autotrophic sulfate reducer joining the already described autotrophic Desulfovibrio sp. strains HRM1, P23 (sharing 88% 16S rRNA gene sequence identity with strain F1), and G11 (sharing 97% 16S rRNA gene sequence identity with strain F1) (McInerney et al. 1981; Brysch et al. 1987; Fichtel et al. 2012). Strain F1 can generate formate and a small amount of acetate from consumption of H2 and CO2. Generation of formate from H2/CO2 by Desulfovibrio strains has been reported previously (Peters et al. 1999), but only chemolithoheterotrophically (i.e., with acetate as fixed carbon source). It was suggested that possessing a formate dehydrogenase is the prerequisite for producing formate during chemolithotrophic growth on H2 (Peters et al. 1999). This is consistent with enzymatic findings of Desulfovibrio desulfuricans which does contain active formate dehydrogenases (Costa et al. 1997). Desulfovibrio sp. strain HRM1 was reported to grow autotrophically (Brysch et al. 1987), but it is not yet known which VFAs are produced during its carbon fixation process. Desulfovibrio sp. strain F1 is thus the first instance of a Desulfovibrio strain reported to produce formate and acetate during autotrophic growth. Formate production was proportional to the initial H2 concentrations in the presence of 2 mM sulfate (~ 0.49 mmol formate per mmol H2) (set H0–H2, Table 2) which is higher than that of Acetobacterium strains (0.083 mmol formate per mmol H2) (Peters et al. 1999). However, formate concentrations increased continually for at least 40 days during growth of Desulfovibrio sp. strain F1, while in the study by Peters et al. formate was only produced transiently (formate concentration peaked before day 20 and then dropped significantly). Moreover, formate production by strain F1 was not completely suppressed when sulfate was as high as 6 mM (Table 2), while Desulfovibrio vulgaris only produced formate under sulfate-limiting conditions (sulfate 0.5 mM) (Peters et al. 1999). These results suggest that the carbon fixation system or gene regulation system in strain F1 may be different from those of previously reported autotrophic sulfate reducers.
Formate is an important intermediate in the acetyl-CoA carbon fixation pathway which is produced in the first step by formate dehydrogenase (Rabus et al. 2006; Müller 2003). Given that strain F1 produced high concentrations of formate, it likely utilizes the acetyl-CoA pathway as the carbon fixing mechanism. The accumulation of formate may be due to the low efficiency of key enzymes in the pathway of forming the methyl branch of acetyl-CoA, which can create a bottleneck for converting formate (Müller 2003). The production of acetate is also intriguing. Previously, autotrophic sulfate reducers utilizing the acetyl-CoA pathway would convert CO2 to acetyl-CoA for anabolism without production of acetate, a key difference from acetogens that gain energy by producing acetate via the acetyl-CoA pathway (Drake et al. 2006). However we show that strain F1, as a sulfate reducer, produces a trace amount of acetate (up to 50 µM) via autotrophic growth. In future we will try to confirm which autotrophic pathway strain F1 utilizes and to identify key functional enzymes for formate/acetate production, for which the genome sequence of strain F1 should be acquired.
Acetate is an important factor when implementing bioaugmentation processes with Dehalococcoides in situ, especially in oligotrophic areas. In this study, we have characterized the acetate utilization profiles by D. mccartyi strain 195. Acetate assimilation is proportional to the final cell density of Dehalococcoides (Fig. 4f), in its role for producing biomass. Consumption of 93 µM acetate was observed when achieving a cell density of 4.8 × 108 mL−1 in strain 195 culture, corresponding to an average of 1.2 × 108 acetate molecules required per cell for the carbon element. The value is close to the calculated value of 1.6 × 108 acetate molecules required per cell for the carbon element, assuming a cell dry weight of 1.2 × 10−14 g per cell (Löffler et al. 2013) and cell chemical composition C5H7O2N. As for the acetate uptake threshold, strain 195 amended with limited acetate decreased the acetate concentration to less than 5 µM. This is consistent with the findings by Wei and Finneran (2013) who found that low acetate concentrations had no limiting effects on TCE dechlorination rates. The lowest acetate concentration tested was 500 µM in Wei and Finneran’s study, while it is 45 µM in this study (Fig. 4). This low acetate utilization threshold is a desirable feature for bioremediation sites, because low concentrations of consistently released acetate sources (either chemical or biological) can be used to provide sufficient fixed carbon to sustain Dehalococcoides while at the same time limiting the growth of other fast-growing acetate-consuming microbes. We observed that although cell growth stopped after acetate was consumed, dechlorination continued as shown in Fig. 4c, d. This was confirmed by the discrepancy between Dehalococcoides cell yields in the mixed culture EC195 (Fig. 1b) and in strain 195 (Fig. 4f), as the former probably suffered from acetate limitation. Continued dechlorination after acetate depletion is expected as acetate does not limit enzymatic activity of reductive dehalogenases which rely on presence of live cells as well as sufficient electron donors and acceptors (H2 and TCE in this case). Although methanogens compete with Dehalococcoides for acetate, they typical utilize acetate down to 7–70 µM (Jetten et al. 1992) while our study demonstrates that Dehalococcoides can use < 5 µM acetate and that a co-culture of Dehalococcoides/Desulfovibrio strain F1 typically accumulated acetate up to only 25 µM. A continuous supply of µM-level acetate by Desulfovibrio thus could favor the growth of Dehalococcoides.
In all, this work suggests that Dehalococcoides species together with autotrophic Desulfovibrio strains could play a significant role in the in situ dechlorination of chloroethenes under lithoautotrophic conditions. Insights gained through the study of this autotrophic dehalogenation system will help to develop more effective engineering approaches for bioremediation of halogenated compounds in the contaminated sites.
References
Abelson PH (1990) Inefficient remediation of ground-water pollution. Science 250:733
Aulenta F, Gossett JM, Papini MP, Rossetti S, Majone M (2005) Comparative study of methanol, butyrate, and hydrogen as electron donors for long-term dechlorination of tetrachloroethene in mixed anerobic cultures. Biotechnol Bioeng 91:743–753
Bramono SE, Lam YS, Ong SL, He J (2011) A mesophilic Clostridium species that produces butanol from monosaccharides and hydrogen from polysaccharides. Bioresour Technol 102:9558–9563
Brysch K, Schneider C, Fuchs G, Widdel F (1987) Lithoautotrophic growth of sulfate-reducing bacteria, and description of Desulfobacterium autotrophicum gen. nov., sp. nov. Arch Microbiol 148:264–274
Chen KY, Morris JC (1972) Kinetics of oxidation of aqueous sulfide by oxygen. Environ Sci Technol 6:529–537
Cheng D, Chow WL, He J (2010) A Dehalococcoides-containing co-culture that dechlorinates tetrachloroethene to trans-1,2-dichloroethene. ISME J 4:88–97
Costa C, Teixeira M, Moura I, LeGall J, Moura JJG, Moura I (1997) Formate dehydrogenase from Desulfovibrio desulfuricans ATCC 27774: Isolation and spectroscopic characterization of the active sites (heme, iron-sulfur centers and molybdenum). J Biol Inorg Chem 2:198–208
Ding C, Chow WL, He J (2013) Isolation of Acetobacterium sp. strain AG that reductively debrominates octa- and penta- brominated diphenyl ether technical mixtures. Appl Environ Microbiol 79:1110–1117
Ding C, Zhao S, He J (2014) A Desulfitobacterium sp. strain PR reductively dechlorinates both 1,1,1-trichloroethane and chloroform. Environ Microbiol 16:3387–3397
Dolfing J, Jiang B, Henstra AM, Stams AJM, Plugge CM (2008) Syntrophic growth on formate: a new microbial niche in anoxic environments. Appl Environ Microbiol 74:6126–6131
Drake H, Küsel K, Matthies C (2006) Acetogenic prokaryotes. In: Dworkin M, Falkow S, Rosenberg E, Schleifer K-H, Stackebrandt E (eds) The Prokaryotes. Springer, New York, pp 354–420
Fichtel K, Mathes F, Könneke M, Cypionka H, Engelen B (2012) Isolation of sulfate-reducing bacteria from sediments above the deep-subseafloor aquifer. Front Microbiol 3:65
Finneran KT, Forbush HM, VanPraagh CVG, Lovley DR (2002) Desulfitobacterium metallireducens sp. nov., an anaerobic bacterium that couples growth to the reduction of metals and humic acids as well as chlorinated compounds. Int J Syst Evol Microbiol 52:1929–1935
Fite A, Macfarlane GT, Cummings JH, Hopkins MJ, Kong SC, Furrie E, Macfarlane S (2004) Identification and quantitation of mucosal and faecal desulfovibrios using real time polymerase chain reaction. Gut 53:523–529
He J, Holmes VF, Lee PK, Alvarez-Cohen L (2007) Influence of vitamin B12 and cocultures on the growth of Dehalococcoides isolates in defined medium. Appl Environ Microbiol 73:2847–2853
He J, Ritalahti KM, Aiello MR, Löffler FE (2003) Complete detoxification of vinyl chloride by an anaerobic enrichment culture and identification of the reductively dechlorinating population as a Dehalococcoides species. Appl Environ Microbiol 69:996–1003
He J, Sung Y, Dollhopf ME, Fathepure BZ, Tiedje JM, Löffler FE (2002) Acetate versus hydrogen as direct electron donors to stimulate the microbial reductive dechlorination process at chloroethene-contaminated sites. Environ Sci Technol 36:3945–3952
Holliger C, Hahn D, Harmsen H, Ludwig W, Schumacher W, Tindall B, Vazquez F, Weiss N, Zehnder AJB (1998) Dehalobacter restrictus gen. nov. and sp. nov., a strictly anaerobic bacterium that reductively dechlorinates tetra- and trichloroethene in an anaerobic respiration. Arch Microbiol 169:313–321
Holmes VF, He J, Lee PKH, Alvarez-Cohen L (2006) Discrimination of multiple Dehalococcoides strains in a trichloroethene enrichment by quantification of their reductive dehalogenase genes. Appl Environ Microbiol 72:5877–5883
Jetten MSM, Stams AJM, Zehnder AJB (1992) Methanogenesis from acetate: a comparison of the acetate metabolism in Methanothrix soehngenii and Methanosarcina spp. FEMS Microbiol Lett 88:181–198
Kaufhold T, Schmidt M, Cichocka D, Nikolausz M, Nijenhuis I (2013) Dehalogenation of diverse halogenated substrates by a highly enriched Dehalococcoides-containing culture derived from the contaminated mega-site in Bitterfeld. FEMS Microbiol Ecol 83:176–188
Krumholz LR, Sharp R, Fishbain S (1996) A freshwater anaerobe coupling acetate oxidation to tetrachloroethene dehalogenation. Appl Environ Microbiol 62:4108–4113
Lane DJ (1991) 16S/23S rRNA sequencing. In: Stackebrandt E, Goodfellow M (eds) Nucleic acid techniques in bacterial systematics. Wiley, New York, pp 115–175
Londry KL, Marais DJD (2003) Stable carbon isotope fractionation by sulfate-reducing bacteria. Appl Environ Microbiol 69:2942–2949
Löffler FE, Yan J, Ritalahti KM, Adrian L, Edwards EA, Konstantinidis KT, Müller JA, Fullerton H, Zinder SH, Spormann AM (2013) Dehalococcoides mccartyi gen. nov., sp. nov., obligately organohalide-respiring anaerobic bacteria relevant to halogen cycling and bioremediation, belong to a novel bacterial class, Dehalococcoidia classis nov., order Dehalococcoidales ord. nov. and family Dehalococcoidaceae fam. nov., within the phylum Chloroflexi. Int J Syst Evol Microbiol 63:625–635
Maymó-Gatell X, Chien YT, Gossett JM, Zinder SH (1997) Isolation of a bacterium that reductively dechlorinates tetrachloroethene to ethene. Science 276:1568–1571
McInerney MJ, Mackie RI, Bryant MP (1981) Syntrophic association of a butyrate-degrading bacterium and methanosarcina enriched from bovine rumen fluid. Appl Environ Microbiol 41:826–828
Mechalas BJ, Rittenberg SC (1960) Energy coupling in Desulfovibrio Desulfuricans. J Bacteriol 80:501–507
Men Y, Feil H, Verberkmoes NC, Shah MB, Johnson DR, Lee PK, West KA, Zinder SH, Andersen GL, Alvarez-Cohen L (2012) Sustainable syntrophic growth of Dehalococcoides ethenogenes strain 195 with Desulfovibrio vulgaris Hildenborough and Methanobacterium congolense: global transcriptomic and proteomic analyses. ISME J 6:410–421
Men Y, Yu K, Bælum J, Gao Y, Tremblay J, Prestat E, Stenuit B, Tringe SG, Jansson J, Zhang T, Alvarez-Cohen L (2017) Metagenomic and metatranscriptomic analyses reveal structure and dynamics of a dechlorinating community containing Dehalococcoides mccartyi and corrinoid-providing microorganisms under cobalamin-limited condition. Appl Environ Microbiol. https://doi.org/10.1128/AEM.03508-16
Muyzer G, Dewaal EC, Uitterlinden AG (1993) Profiling of complex microbial populations by denaturing gradient gel electrophoresis analysis of polymerase chain reaction-amplified genes coding for 16S rRNA. Appl Environ Microbiol 59:695–700
Muyzer G, Stams AJM (2008) The ecology and biotechnology of sulphate-reducing bacteria. Nat Rev Microbiol 6:441–454
Müller V (2003) Energy conservation in acetogenic bacteria. Appl Environ Microbiol 69:6345–6353
Peters V, Janssen PH, Conrad R (1999) Transient production of formate during chemolithotrophic growth of anaerobic microorganisms on hydrogen. Curr Microbiol 38:285–289
Rabus R, Hansen TA, Widdel F (2006) Dissimilatory sulfate- and sulfur-reducing prokaryotes. Prokaryotes 2:659–768
Ramsburg CA, Abriola LM, Pennell KD, Löffler FE, Gamache M, Amos BK, Petrovskis EA (2004) Stimulated microbial reductive dechlorination following surfactant treatment at the Bachman Road site. Environ Sci Technol 38:5902–5914
Richardson RE, Bhupathiraju VK, Song DL, Goulet TA, Alvarez-Cohen L (2002) Phylogenetic characterization of microbial communities that reductively dechlorinate TCE based upon a combination of molecular techniques. Environ Sci Technol 36:2652–2662
Ritalahti KM, Löffler FE (2004) Populations implicated in anaerobic reductive dechlorination of 1,2-dichloropropane in highly enriched bacterial communities. Appl Environ Microbiol 70:4088–4095
Schink B (1997) Energetics of syntrophic cooperation in methanogenic degradation. Microbiol Mol Biol Rev 61:262–280
Schneidewind U, Haest PJ, Atashgahi S, Maphosa F, Hamonts K, Maesen M, Calderer M, Seuntjens P, Smidt H, Springael D, Dejonghe W (2014) Kinetics of dechlorination by Dehalococcoides mccartyi using different carbon sources. J Contam Hydrol 157:25–36
Scholz-Muramatsu H, Neumann A, Meßmer M, Moore E, Diekert G (1995) Isolation and characterization of Dehalospirillum multivorans gen. nov., sp. nov., a tetrachloroethene-utilizing, strictly anaerobic bacterium. Arch Microbiol 163:48–56
Sharma PK, McCarty PL (1996) Isolation and characterization of a facultatively aerobic bacterium that reductively dehalogenates tetrachloroethene to cis-1,2-dichloroethene. Appl Environ Microbiol 62:761–765
Wei N, Finneran KT (2013) Low and high acetate amendments are equally as effective at promoting complete dechlorination of trichloroethylene (TCE). Biodegradation 24:413–425
Acknowledgements
This work was supported by Ng Teng Fong Charitable Foundation (NTFCF) Fund [Grant Number R302-000-198-720] and the National Research Foundation, Prime Minister’s Office, Singapore under the Competitive Research Programme [Grant Number NRF-CRP5-2009-05]. We thank Professor Stephen H. Zinder at Cornell University for providing D. mccartyi strain 195.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no competing financial interest.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Ding, C., Alvarez-Cohen, L. & He, J. Growth of Dehalococcoides mccartyi species in an autotrophic consortium producing limited acetate. Biodegradation 29, 487–498 (2018). https://doi.org/10.1007/s10532-018-9846-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10532-018-9846-9