
Abstract
There is growing evidence for the decline of cryptic species across the planet as a result of human activities. Accurate data regarding patterns of poorly known or hard-to-find species diversity is essential for the recognition and conservation of threatened species and ecosystems. Casey’s June beetle Dinacoma caseyi is a federally listed endangered species restricted to the Coachella Valley in southern California, where rapid development is leading to habitat fragmentation. This fragmentation may be disproportionately impacting a wide-range of poorly-dispersing, cryptic species, including Casey’s June beetle, which has flightless females. We characterized 1876 single nucleotide polymorphisms from across the genome along with 1480 bp of mitochondrial DNA of all confirmed extant Dinacoma populations. We found that Dinacoma is isolated into three distinct species, including evidence for a previously undescribed species revealed during this study. Each is restricted to a small part of the inland desert region. Our results suggest unappreciated and fine scale diversity, which may be reflected in the other cryptic species of the region. Patterns of diversity in non-vagile species should guide ongoing conservation planning in the region. These results show that genetic exchange within the one remaining island of beetle habitat (Palm Canyon Wash) is not limited. However, non-vagile species, such as Casey’s June beetle, may not colonize suitable but fragmented habitat islands which presents risks to the species due to habitat loss and periodic natural events that may put the single population at risk of extirpation.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Although insects represent the majority of Earth’s known and predicted biodiversity (Mora et al. 2011), they receive a disproportionately small fraction of conservation resources. There is overwhelming, growing evidence that many insect communities are in decline, and that these insect communities may be at least as vulnerable as associated vertebrate communities (Vogel 2017; Lister and Garcia 2018; Loboda et al. 2018; Sánchez-Bayo and Wyckhuys 2019). Yet with the majority of conservation resources dedicated to vertebrates, the implicit assumption is that habitat conservation plans relying on vertebrate umbrella species will preserve invertebrates, despite evidence to the contrary (e.g. Rubinoff 2001; Rubinoff and Sperling 2004; Régnier et al. 2009, 2015; Mckinney 1999). Conservation planning that continues to ignore threatened and less vagile invertebrates may leave them and similar species in peril, threatening biodiversity and ecosystem integrity. While it is not practical to intensively survey all threatened invertebrate populations, using appropriate invertebrate surrogates that reflect the population and landscape-scale challenges faced by less dispersive organisms will contribute to a more realistic and effective template for inclusive reserve planning and conservation of biodiversity.
California is a biodiversity hotspot and the subject of extensive conservation planning and development mitigation schemes for decades (e.g. CDFG 1993; CVAG 2016); fine scale patterns of endemism make it an ideal region for investigating how cryptic invertebrate diversity might be used to improve ongoing reserve design. The phylogeography of California species is complex, and variations in geology and microclimates have helped to generate the largest number of endemic species of any U.S. state (Stein et al. 2000); unfortunately, California also hosts more endangered animals than anywhere else in the nation (Dobson et al. 1997; USFWS 2019). In southern California, much of the conservation attention has focused on the coastal region which supports the highest levels of regional endemism (Stein et al. 2000) and development due to the expansion of the Los Angeles-San Diego metropolitan corridor. In recent decades, urbanization has pushed further east, and the desert valley regions are undergoing rapid conversion. The Coachella Valley, just east of Los Angeles, now hosts multiple federally listed endangered species prompting the creation of a Multiple Species Habitat Conservation Plan to balance human needs with the conservation of remaining natural areas critical to preserving endangered biological diversity (Alagona and Pincetl 2008). As usual, the focus has been on using vertebrate surrogates to conserve less-visible species. Conversely, little has been done to assess patterns of diversity for rare invertebrates and how they might serve as useful umbrella species for vertebrates and other, less vagile and more cryptic species that are also threatened by habitat destruction.
Unfortunately, data collection reflecting endangered invertebrate diversity and endemism in these inland desert areas has not kept up with the pace of development, possibly eliminating restricted species that needed conservation attention. A failure to recognize unique genetic diversity can lead to mismanagement and unnecessary extinction, since small populations with limited genetic exchange typically experience a loss of genetic diversity and face higher extinction risk (Avise 1989; Frankham 2005). Incomplete knowledge of species boundaries and cryptic diversity can impart an erroneously optimistic impression of connectivity among what are actually disjointed populations, or even taxa that represent distinct species or sub-species (Keogh et al. 2008; Murphy et al. 2011). Without such crucial information, effective conservation of diversity is difficult or impossible, undermining urgently needed action while squandering resources applied to ill-informed mitigation. Phylogeographic research focused on fine-scale patterns of biodiversity and species boundaries are essential for effective conservation, and such intensive studies often reveal novel, cryptic species diversity (Holland and Hadfield 2002; Shaffer et al. 2004; Griffiths et al. 2010).
As a result of rapid development in the Coachella Valley, the region’s biota is in urgent need of phylogeographic studies to better document patterns of diversity and endemism, and guide conservation strategies. Casey’s June beetle (Dinacoma caseyi Blaisdell 1930) is a federally listed endangered species, restricted to the vicinity of Palm Springs, the largest city in the Coachella Valley; it has suffered dramatic population reductions due to development of its desert habitat (USFWS 2011). It is one of two described members of a genus confined to particular alluvial habitats in southern California, both species with flightless, fossorial, females occurring in highly localized populations. As such, the beetle represents a conservative model for understanding the impacts of fragmentation on population connectivity and isolation of the Coachella Valley’s less vagile endemic species. Additionally, the genetic independence of D. caseyi and its sole congener, D. marginata, which occurs in isolated pockets to the south and west of D. caseyi, has never been phylogenetically examined and may be important in understanding broader patterns of biodiversity in other cryptic species in the region. While historically more widespread on the coast, Dinacoma now appears to be largely restricted to three isolated populations. Thus, the genus Dinacoma presents an ideal opportunity to investigate both local and regional patterns of cryptic species endemism among widely isolated populations in varied ecological contexts in southern California.
Nothing is known about systematic and genetic relationships between Dinacoma species, and both fine-scale and broader geographic assessments of relationships are needed. Further, since the taxonomy of Dinacoma has never been investigated at the molecular level, we also wanted to assess DNA-based support for the recognition of any unique populations within D. caseyi and and species-level divisions across Dinacoma, and with respect to populations of the sister species D. marginata. We have sampled the full range of D. caseyi. D. marginata was described from coastal southern California but hasn’t been recollected there in decades (likely due to obliteration of habitat). The other two populations we used, Bautista Canyon and San Felipe, are either newly discovered (San Felipe) or the only known source currently available (Bautista) for D. marginata. There may be other, unconfirmed, populations of Dinacoma isolated in pockets of southern California, or even Baja California, Mexico, but they are rare and were not available for this study.
Characterization of cryptic diversity and structure among Dinacoma populations and species is not only vital for management of D. caseyi, but, because the females are fossorial and flightless, also provides insight into broader phylogeographic patterns of endemism that may be shared by other poor-dispersing taxa in the threatened desert regions of southern California. Because mtDNA has historically been used to assess species and conservation status for a wide range of animals (e.g. Moritz 1994; Rubinoff 2006), comparing its utility to more data-rich but costly genomic techniques is relevant to future conservation genetic research. Dinacoma also provides an opportunity to understand how patterns of inheritance in the maternally transmitted mtDNA genome might be disproportionately impacted by extremely biased sex-based dispersal (in this case flightless females) as compared to patterns from nuclear markers. We used a combination of Sanger sequencing and double-digest restriction-site associated DNA sequencing (ddRAD) to investigate the population genetics, biogeography, and phylogenomics of Dinacoma and asked the following questions: Is there evidence for the reciprocal monophyly of the Dinacoma species and populations or do they show signs of recent genetic exchange? What is the genetic diversity and distribution of the endangered D. caseyi as compared to Dinacoma populations outside of the Palm Springs area? How is development in the Coachella Valley affecting fine scale genetic diversity in D. caseyi, as reflected by genomic structuring? Has having flightless females led to more genetic isolation in maternally inherited mtDNA than nDNA across populations? And, what are the phylogeographic patterns in Dinacoma as they relate to current and future conservation planning? While this study is focused on a single genus, the focal species is a federally listed endangered species and, by virtue of its cryptic life history and flightless females, likely represents a more sensitive model species for habitat conservation plans that are intended to save a broader measure of biodiversity. By focusing on the most sensitive, rather than the more resilient endemic members of a threatened ecosystem, conservationists might have a better chance to preserve overall community structure and function.
Methods
We used both genomic sampling of genome-wide nuclear DNA and 1480 bp of mitochondrial (mt) DNA data from cytochrome oxidase subunit I (COI) to understand population structure and diversity within the remnant Dinacoma caseyi population. By comparing nuclear and mitochondrial datasets, we can not only explore the impacts of urbanization and inform conservation efforts for this recently listed, endangered beetle, but also examine the relative sensitivity and utility of nuclear vs. mitochondrial markers.
Sample acquisition and DNA extraction
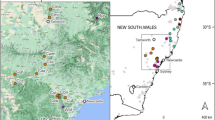
Twenty adult D. caseyi and 28 D. marginata from three localities representing much of the range of the genus (Fig. 1) were collected into 70% + EtOH. Dinacoma caseyi were collected by hand at blacklights from their only known range in Palm Springs, Riverside County, CA. Dinacoma marginata were collected at blacklights from the two known extant population sites representing divergent parts of their inland range, from the San Felipe Valley in San Diego County, CA in the south, north to the Bautista Canyon region near Hemet in Riverside County, CA, only 40 km from where D. caseyi occurs. Two legs were dissected from adult beetles for DNA extraction. The remainder of each sample was placed into fresh 90–95% EtOH in separate, labeled, vials and deposited in the University of Hawaii Insect Museum (UHIM) as a voucher specimen at − 80 °C.

Dinacoma sampling map. Colored markers represent collection localities from this study. D. caseyi all collected from the Palm Springs/Cathedral City area; D. marginata collected from Bautista Canyon near Hemet, CA (northern point) and the San Felipe Valley region (southern point). Grey markers indicate additional historic collection points for members of the Dinacoma genus. Dots encompass all known collection localities for Dinacoma
Separate DNA extractions were prepared for Sanger sequencing and for ddRAD in the labs of DR and SG, respectively. For Sanger sequencing, DNA was extracted from a single leg using the DNeasyTM Blood & Tissue kit (Qiagen). Tissue was manually ground in 1.5 mL Eppendorf safe-lock tubes (Eppendorf North America, Hauppauge, NY) using tube-fitted pestles. Proteinase-K digestion enzyme was added to tissue followed by 24 h incubation at 55 °C. During this time, samples were constantly rotated using a VWR Tube Rotator (Avantor, Radnor, PA). All remaining extraction protocols followed Qiagen standards. For ddRAD, a single leg was homogenized in tissue lysis buffer using a 2010 Geno/Grinder Automated Tissue Homogenizer and Cell Lyser (SPEX SamplePrep, Metuchen, NJ, USA) for 30 s at 1500 rpm. The homogenate was then incubated in a 55 °C water bath for 3 h. Incubation was followed by extraction on a Kingfisher Flex 96 automated extraction instrument (Thermo Scientific, Waltham, MA) using standard protocols with a NucleoMag Tissue Kit (MACHEREY-NAGEL, Düren, Germany). The quantity and quality of the extracted DNA samples were determined using a Quant-it Picogreen assay (Thermo Fisher, Waltham, MA) on a SpectraMax M2 Plate Reader (Molecular Devices, Sunnyvale, CA).
Sanger sequencing
Polymerase chain reaction (PCR) for COI was performed using either a BioRad T100™ or C1000 Touch™ thermal cycler. Primer pairs LCO-1490/HCO-2198 and Jerry/Pat2 were used to sequence 1480 base pairs of the COI (Folmer et al. 1994; Simon et al. 1994) under the following thermal regime: 3 min at 94 °C, 40 cycles of 94 °C for 30 s, 50 °C for 30 s, and 70 °C for 1 min, followed by a final 70 °C extension for 10 min and a 4 °C hold until termination. For those samples where both primer pairs failed to amplify, we paired primers LCO-1490/Pat2 to limit the potential for binding errors along the amplicon. This proved effective for a substantial portion of otherwise difficult samples. PCR products were purified using QIAquick® spin columns (Qiagen) following standard protocols. Sanger sequencing services were provided by either Eurofins Genomics (www.eurofinsgenomics.com) or the ASGPB Laboratory at the University of Hawaii Manoa (www.hawaii.edu/microbiology/asgpb). All sequences were aligned and manually checked for errors using geneious v7.1.9 (https://www.geneious.com, Kearse et al. 2012). Alignment was completed using the Geneious aligner algorithm with default parameters. Protein translation was conducted to identify a suitable reading frame and survey for internal stop codons signaling potential pseudogenes. To identify an outgroup, we found the most closely related Scarab from the melolonthine subfamily using COI sequence data from the Barcoding of Life Database (BOLD), which was Amblonoxia fieldi, a species that occurs broadly in the southwestern U.S. and is found along the southern coast of California. Amblonoxia fieldi sequences were used to root analyses of COI data. Amblonoxia and Dinacoma are 2 of 11 genera within the tribe Melolonthini, including the more widespread genera Polyphylla and Phyllophaga. Unfortunately, no studies have included sampling of Dinacoma and Amblonoxia, however from preliminary analyses of all available sequences on BOLD, they appear to be more closely related to each other than any of the other genera in the tribe.
ddRAD library preparation
ddRAD DNA extractions were normalized to 4 ng/µL in 44.5 µL dH2O and ddRAD libraries were prepared following Peterson et al. (2012). To prepare the library, 175 ng of DNA from each individual was digested using the restriction enzymes NlaIII and MluCI. One of 48 unique barcode adapters was ligated to the restriction overhang, generating inline barcodes. Two subpools of samples containing 28 and 29 of these barcodes were generated and size-selected using a 1.5% agarose gel cassette on a Blue Pippin electrophoresis unit (Sage Science, Beverely, MA) with a target size selection of “narrow 400 bp”. The final PCR amplification step was run for 10 cycles, during which Illumina i7 barcodes were added for each sub-pool, and PCR products were cleaned using solid-phase reversible immobilization (SPRI) beads at a 1.5:1 ratio of PEG containing bead solution to sample volume (DeAngelis et al. 1995; Rohland and Reich 2012). The cleaned subpool libraries were analyzed for quantity and size distribution using the NGS Fragment Analysis Kit on a Fragment Analyzer and pooled at equal molar ratios to generate the final library (containing 57 individuals). This library was sequenced on a single lane of 100 bp single-end sequencing on an Illumina HiSeq 4000 sequencer.
Read clustering and SNP selection
Raw Illumina sequencing reads were processed using the stacks v. 2.1 (Catchen et al. 2013) pipeline. In stacks, the process_radtags function was used to clean and demultiplex data, removing reads with uncalled bases or low-quality scores and rescuing barcodes when possible. Given the lack of a reference genome for Dinacoma, de novo assembly of loci was conducted using the denovo_map.pl wrapper. The program was parameterized to allow 2 mismatches between loci within individuals and 3 mismatches between loci between individuals when creating the catalog, and other parameter set at default values. The stacks core program populations was used for filtering and final SNP calling. The first SNP at each locus was retained, and SNPs found in < 15% of individuals were removed. The resulting dataset was then inspected, and a missing data threshold of 20% was used to identify and remove individuals with low quality data; the above procedures were then repeated without the low-quality individuals. The final dataset was subjected to additional filtering using vcftools to exclude sites with greater than 25% missing data (Danecek et al. 2011). geneious v. 10.2.4 (Kearse et al. 2012) and pgdspider v. 2.1.1.2 (Lischer and Excoffier 2012) were used to convert data between different file formats. To facilitate fine-scale investigations for Dinacoma populations and minimize intraspecific missing data and singleton/monomorphic SNPs, we also used these procedures to create three datasets using only individuals collected from one of the three sampled populations (Palm Springs in Riverside County, n = 16; San Felipe Valley (SF) in San Diego County, n = 12; and Bautista Canyon (BC) near Hemet in Riverside County, n = 14).
Population genetics and phylogenetics
We calculated descriptive statistics and pairwise Nei’s Gst for the COI dataset using pegas v0.12 (Paradis 2010) and strataG v2.0.2 (Archer et al. 2017), and mmod v1.3.3 (Winter 2012), respectively, in R v3.6.2 (R Core Team 2015). We assessed population structure with phylogenetic likelihood using raxml (Stamatakis 2014). Analyses with COI mitochondrial data were completed using the RAxML-HPC2 Workflow on XSEDE implemented on the CIPRES Science Gateway (Miller et al. 2010) running raxml v. 8.2.10. Analyses were parameterized to perform 25 alternative runs on distinct starting trees including bootstrapping calculations halted based on autoMRE criterion. Remaining parameters were left on default settings. Sanger data was used to construct two maximum-likelihood phylogenetic trees in raxml: one that makes use of full sequences and a second with missing data from 5′ and 3′ ends removed (approximately 407 base pairs/individual retained). A COI haplotype network was also constructed for mitochondrial data in PopART (https://popart.otago.ac.nz) using TCS network inference (Clement et al. 2002).
Descriptive population genetic statistics for the ddRAD dataset were calculated in GenoDive v3.0.3 (Meirmans and Van Tienderen 2004). We also used GenoDive to calculate pairwise population differentiation based on G″ST using 10,000 replicates, and test for deviations from Hardy–Weinberg equilibrium using 10,000 permutations of the least-squares method. The complete ddRAD data set (all individuals) was used to create a single, unrooted likelihood tree in raxml with the same procedure used for mitochondrial data, except with the addition of a Lewis ascertainment bias correction (Lewis 2001). Lewis ascertainment bias correction requires that potentially invariable sites are removed from the data set. Removal was completed in r v. 3.4.1 (R Core Team 2015) with a custom script employing the phrynomics v. 2.0 (Banbury and Leache 2014) and phylotools v. 0.1.2 (Zhang et al. 2012) packages. We also completed structure analyses to assess structure using a Bayesian clustering criterion (Pritchard et al. 2000). Using structure v. 2.3.4, we ran one hundred simulations for each K value 1 to 15 with 150,000 repetitions and a 50,000-repetition burn-in under an admixture model with correlated allele frequencies among populations. Unspecified parameters were kept default. Evanno’s ΔK (Evanno et al. 2005) and LnPr(K|X) (Pritchard et al. 2000) were used to determine the optimum number of clusters for the data. CLUMPP was used to merge runs and generate a consensus plot (Jakobsson and Rosenberg 2007). The pophelper v. 2.2.1 (Francis 2017) package employed in r v. 3.4.1 was used to implement Evanno’s method and CLUMPP, and to generate the structure barplot. We also used the clumpak web portal (clumpak.tau.ac.il/index.html Kopelman et al. 2015) to produce and inspect bar plots across the entire range of K-values (K = 1–15) (Fig. S1).
Both principle component analysis (PCA) and Neighbor-Joining analysis (Saitou and Nei 1987) were conducted for the ddRAD dataset in r v. 3.4.1 and SplitsTree4 v. 4.14.5 (Huson and Bryant 2006), respectively (Figs. S2, S3). Principal component analysis in r was completed using a custom script employing the package SNPrelate v. 1.16.0 (Zheng et al. 2012).
Within population substructure
Within-population substructure was assessed further using the locality-specific ddRAD datasets. For each population, an unrooted likelihood tree was constructed in raxml following the above procedures used for the complete ddRAD dataset tree. We completed structure analyses on these datasets using the previously described procedures with K values 1–6.
Results
Data properties
Sanger sequencing of the 1480 base pair, mtDNA amplicon was successful for 38 of 48 Dinacoma samples (see metadata). Amblonoxia fieldi COI barcode sequences from two individuals were used to root COI trees constructed in raxml. Illumina sequencing generated 320,474,796 raw reads, with an average read count of 5,622,365 per individual. stacks assembled raw reads into 4,630,270 catalog loci. Six of the original 48 Dinacoma samples (4 D. caseyi; 2 D. marginata) were low quality and excluded from our ddRAD analyses based on the percent-missing-data threshold. The final filtered dataset contained 699 SNPs for all individuals (n = 42), and the population-restricted datasets for Palm Springs (n = 16), Bautista Canyon (n = 14) and the San Felipe Valley (n = 12) samples contained 1568, 1253 and 1876 SNPs, respectively.
Sanger sequencing
We observed relatively low nucleotide and haplotype diversity in the COI dataset, ranging from 0.0017 to 0.0025 and 0.564 to 0.833, respectively (Table 1). Considering three “populations” (see below for rationale), estimates of Tajima’s D were negative for D. marginata in the San Felipe Valley and positive for the other two populations, although none of these estimates were statistically significant (Table 1). The two maximum likelihood phylogenies generated using COI data had identical topologies; the tree generated from all available sequence data is displayed in Fig. 2. The maximum likelihood phylogeny divided Dinacoma into three distinct clades: one representing D. caseyi from the Palm Springs region, and two representing D. marginata (Fig. 2). Within the marginata complex, samples from Bautista Canyon form a distinct clade from those from San Felipe Valley. The BC D. marginata clade is basal to the D. caseyi and SF D. marginata, clade, and this pattern was supported by pairwise distance measures of the COI dataset (Table 2). However, despite these geographic patterns, marginal to low bootstrap support values (> 80%) were obtained for these clades. The TCS haplotype network (Fig. S4) supports a clear separation of D. caseyi and two distinct clades of D. marginata reflecting the isolation of populations at different collection localities.

Maximum Likelihood tree generated by raxml using COI sequence data and public COI sequences available on BOLD for A. fieldi outgroup. Outgroup sequences of varying length nested within the 1480 bp amplicon targeted by this study. ML bootstrap values at corresponding nodes
ddRAD sequencing
Measures of observed and expected heterozygosity and inbreeding from the ddRAD dataset were relatively uniform across populations, ranging from 0.045 to 0.083, 0.05 to 0.094, and 0.089 to 0.123, respectively (Table 3). Dinacoma marginata from Bautista Canyon displayed the highest heterozygosity and inbreeding compared to the other populations. Thirty-five loci of the 699 were observed to deviate from HWE, however descriptive statistics changed little when these loci were removed (Table 3), so we considered the entire dataset of 699 SNPs for downstream analyses. The unrooted maximum likelihood phylogeny generated by raxml clearly distinguishes the three Dinacoma clades identified with COI data with high bootstrap support (Fig. 3). Clades appear to be evolutionarily equidistant from each other, or nearly so. While we lacked an outgroup to root the ddRAD phylogeny, pairwise differentiation measures of this dataset indicated a closer relationship between the two D. marginata populations, than compared to the D. caseyi population (Table 2). Analysis of structure runs using ΔK and LnPr(K|X) supported K = 3 as the optimal cluster number (Figs. 4, S5). These three clusters correspond to D. caseyi from its range in the Palm Springs/Cathedral City area, BC D. marginata, and SF D. marginata and agree with the clades identified by raxml (Fig. 4).

Base tree is a maximum likelihood tree generated in raxml for the stacks dataset including all sampled individuals. ML Bootstrap support values displayed. Weak support (< 60) was obtained for all within-clade branching, with most receiving support values < 30. Scale bar indicates mean substitutions per site. Boxes contain unrooted population specific raxml trees for each of the localities sampled. All branches receive weak support (< 60) signifying an overall lack of within population substructure

Bar plot summarizing merged results for 100 structure runs at the determined cluster optimum, K = 3
We looked for substructure in the mitochondrial haplotype network and through the use of hierarchical raxml and structure analyses for our genomic data, but found none. Figure 3 shows ML trees from population specific datasets with no branches having high support. Hierarchical structure analyses all support K = 1 and are provided in the supplemental materials (Fig. S5). Descriptive statistics generated from the region-specific ddRAD datasets unsurprisingly showed higher heterozygosity estimates compared to the dataset generated from all populations (Table 3). Estimates of inbreeding were higher for D. marginata from Bautista Canyon, but lower in the other two populations (Table 3).
Discussion and conclusions
Our results strongly support the species-status of D. caseyi as a distinct, diagnosable, and isolated taxon from D. marginata. Further, our results strongly suggest the existence of an undescribed species of Dinacoma from San Felipe Valley which is equally isolated from the typical D. marginata population we sampled from Bautista Canyon. These results not only support the continued conservation of D. caseyi as a unique and isolated species, but also reveal a broader pattern of isolation among Dinacoma lineages. Because the isolation is supported by both the mtDNA and the more rapidly evolving nDNA from the ddRAD data, it suggests that there is very little, if any, active genetic exchange – even by the flighted male beetles – between these species. Estimates of inbreeding in these lineages also supported this conclusion. Conservation planning for the Coachella Valley should not assume that other poorly-known species all represent singular entities simply because they share a species name. Our results suggest that, for many groups, the region may represent a mosaic of unique, endemic species, and that additional assessment of cryptic species diversity is needed before conservation reserve planning proceeds further than it already has. If Multiple Species Habitat Conservation Plans continue to be designed with the assumption that most species occur region-wide, our results suggest endemic species will be lost.
Dinacoma’s flightless females make the isolation of mtDNA across the region perhaps unsurprising, but the genomic nDNA data confirms that the males, which disperse through evening flights, are not traveling across the region between Bautista Canyon, San Felipe Canyon and the Palm Springs area. This suggests that other groups, even those with flight, may be naturally isolated into pockets of endemic species, worthy of conservation attention. Yet, within each population of Dinacoma, including D. caseyi, neither mtDNA nor nDNA show sub-structure. For D. caseyi, in particular, our sampling across the known distribution included many sites recently isolated by development, which was feared to be isolating this endangered species into unsustainable population fragments since roads are known dispersal barriers, even to flighted insects (Andersson et al. 2017; Muñoz et al. 2015). Our results provide indirect evidence for two important management phenomena in this regard. Firstly, nDNA indicates that not only are males flying freely within the D. caseyi population, but also that females are maintaining mtDNA genetic contact at some level, despite being flightless. Alternatively, this lack of local isolation may suggest that the habitat destruction and resultant isolation suffered by D. caseyi is too recent to reflect now-isolated groups of females on fragments of remaining habitat. Additional research to understand the dispersive abilities of female D. caseyi will be important in understanding the impact of roads and urbanization on population connectivity.
Evaluation of population fragmentation and inbreeding
One of the most important findings from this study is confirmation that D. caseyi is a distinct species, diagnosable from D. marginata. This not only justifies its protected status, but also reflects finer scale biodiversity patterns across a highly heterogenous inland desert region. Within D. caseyi, there was no detectable structure in the population, even between samples from locations separated by housing developments and highways, possibly due to the recent nature of habitat fragmentation as discussed above. We suspect that COI is evolving too slowly to reflect the isolation that development has caused between females in the past few decades, and that the flighted males are dispersing between patches of habitat, reflecting the more sensitive genomic (nDNA) dataset’s lack of sub-structuring. Effective conservation management planning may still involve establishing habitat connectivity between D. caseyi populations to ensure long term genetic exchange as has been suggested for preserving insect diversity in general (Samways 2007) and beetles specifically (Eggers et al. 2010). Estimates of inbreeding generated from the ddRAD dataset indicated that individuals in all populations were more related to each other than would be expected with random mating (i.e. positive GIS). While D. caseyi appeared to have less inbreeding compared to D. marginata (Table 3), our limited sample sizes preclude strong comparative conclusions between populations of this species.
Evidence of deep isolation
A surprising result of this work is the robust support for the recognition of a new species of Dinacoma. The genomic divergence between the BC and SF populations is equivalent to that between D. caseyi and either D. marginata population, reflecting relatively deep divergence across the genus. Since the BC population is already recognized as D. marginata and is geographically closest to the type locality for D. marginata in coastal San Diego county, we continue to recognize BC as D. marginata. Unfortunately, the type locality is now heavily developed and Dinacoma has not been collected from the exact area in decades. Regardless of the taxonomy, the salient message from the data is that all three Dinacoma populations represent species-level divergence. This divergence is supported not only by mtDNA, which reflects the isolation of the flightless females, but also by the nDNA exchanged by the dispersive males. Isolation between Dinacoma species suggests the possibility of much finer scale patterns of biodiversity and speciation in a variety of other cryptic species across what might appear to be contiguous, homogenous and rapidly developing desert areas of southern California. Effective multi-species conservation planning should not assume that genetic exchange is occurring in what are currently considered widespread species in other cryptic groups. Each subregion may need to make independent investigations to save local and irreplaceable lineages that represent an unexpectedly rich and complex history of biodiversity; cryptic species diversity is essential in this regard (Bickford et al. 2007).
The cryptic nature of Dinacoma species presents a fascinating but ominous narrative for the future of many endemic species in southern California’s western deserts. If Dinacoma is typical, and there is no reason to assume it is not, there are many, highly restricted species exhibiting fine scale patterns of speciation and isolation. The addition of genomic data to past and current studies will help reveal this hidden diversity. Unfortunately, efforts to identify and understand cryptic species are often neglected (Trontelj and Fišer 2009) in favor of management centered around charismatic taxa (eg. Rubinoff 2001). The implicit assumption is that they share similar diversification patterns and so current conservation plans will be adequate for the whole of the regional biota. Our findings suggest that this assumption is not accurate. In practice, a cryptic, sessile genus such as Dinacoma likely represents a more sensitive and detailed conservation model for the southern California desert community. Landscape-level changes have already irreparably reduced the span of the natural areas remaining in the region and may have extirpated species without them ever coming to light. We emphasize that the western deserts of southern California merit more intensive and careful biotic surveying to accommodate human use with as much endemic biodiversity as possible.
Data availability
Raw ddRAD reads are available on NCBI. BioProject: PRJNA552048; SRA: SRR9646661-SRR9646708. Sanger sequencing data is available on BOLD and GenBank MT324554-MT324591.
References
Alagona PS, Pincetl S (2008) The Coachella valley multiple species habitat conservation plan: a decade of delays. Environ Manag. https://doi.org/10.1007/s00267-007-9018-x
Andersson P, Koffman A, Sjödin NE, Johansson V (2017) Roads may act as barriers to flying insects: species composition of bees and wasps differs on two sides of a large highway. Nat Conserv 18:47
Archer FI, Adams PE, Schneiders BB (2017) stratag: an R package for manipulating, summarizing and analysing population genetic data. Mol Ecol Resour 17:5–11
Avise JC (1989) A role for molecular genetics in the recognition and conservation of endangered species. Trends Ecol Evol 4(9):279–281
Banbury B, Leache A (2014) Phrynomics: an SNP toolkit. R package version 2.0.
Blaisdell FE (1930) Revision of the genus and species of dinacoma with description of a new species (Coleoptera: Scarabaeidae). Pan Pac Entomol 6(4):171–177
Bickford D, Lohman DJ, Sodhi NS, Ng PK, Meier R, Winker K, Ingram KK, Das I (2007) Cryptic species as a window on diversity and conservation. Trends Ecol Evol 22(3):148–155
California Department of Fish and Game (1993) Southern California coastal sage scrub natural communities conservation plan. Scientific review panel conservation guidelines and documentation. California Department of Fish and Game, Sacramento, California
Catchen J, Hohenlohe PA, Bassham S, Amores A, Cresko WA (2013) Stacks: an analysis tool set for population genomics. Mol Ecol 22(11):3124–3140
Clement M, Snell Q, Walker P, Posada D, Crandall K (2002) TCS: estimating gene genealogies. Parallel and distributed processing symposium, international proceedings 2:184
Coachella Valley Association of Governments (2016) Coachella valley multiple species habitat conservation plan. CVMSHCP plan documents. Coachella Valley Association of Governments, Palm Desert, California. https://www.cvmshcp.org
Danecek P, Auton A, Abecasis G, Albers CA, Banks E, DePristo MA et al (2011) The variant call format and VCFtools. Bioinformatics 27(15):2156–2158
DeAngelis MM, Wang DG, Hawkins TL (1995) Solid-phase reversible immobilization for the isolation of PCR products. Nucleic Acids Res 23(22):4742
Dobson AP, Rodriguez JP, Roberts WM, Wilcove DS (1997) Geographic distribution of endangered species in the United States. Science 275(5299):550–553
Eggers B, Matern A, Drees C, Eggers J, Haerdtle W, Assmann T (2010) Value of semi-open corridors for simultaneously connecting open and wooded habitats: a case study with ground beetles. Conserv Biol 24(1):256–266
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14(8):2611–2620. https://doi.org/10.1111/j.1365-294X.2005.02553.x
Folmer O, Black M, Hoeh W, Lutz R, Vrijenhoek R (1994) DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates. Mol Mar Biol Biotech 3(5):294–299
Francis RM (2017) Pophelper: an R package and web app to analyse and visualize population structure. Mol Ecol Resour 17(1):27–32. https://doi.org/10.1111/1755-0998.12509
Frankham R (2005) Genetics and extinction. Biol Conserv 126(2):131–140
Griffiths AM, Sims DW, Cotterell SP, El Nagar A, Ellis JR, Lynghammar A, Serra-Pereira B et al (2010) Molecular markers reveal spatially segregated cryptic species in a critically endangered fish, the common skate (Dipturus batis). Proc R Soc Lond B 277(1687):1497–1503
Holland BS, Hadfield MG (2002) Islands within an island: phylogeography and conservation genetics of the endangered Hawaiian tree snail Achatinella mustelina. Mol Ecol 11(3):365–375
Huson DH, Bryant D (2006) Application of phylogenetic networks in evolutionary studies. Mol Biol Evol 23(2):254–267. Software available from www.splitstree.org
Jakobsson M, Rosenberg NA (2007) CLUMPP: a cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics 23(14):1801–1806. https://doi.org/10.1093/bioinformatics/btm233
Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S et al (2012) Geneious basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28(12):1647–1649
Keogh JS, Edwards DL, Fisher RN, Harlow PS (2008) Molecular and morphological analysis of the critically endangered Fijian iguanas reveals cryptic diversity and a complex biogeographic history. Philos Trans R Soc B 363(1508):3413–3426
Kopelman NM, Mayzel J, Jakobsson M, Rosenberg NA, Mayrose I (2015) Clumpak: a program for identifying clustering modes and packaging population structure inferences across K. Mol Ecol Resour 15(5):1179–1191
Lewis PO (2001) A likelihood approach to estimating phylogeny from discrete morphological character data. Syst Biol 50(6):913–925. https://doi.org/10.1080/106351501753462876
Lischer HE, Excoffier L (2012) PGDSpider: an automated data conversion tool for connecting population genetics and genomics programs. Bioinformatics 28(2):298–299
Lister BC, Garcia A (2018) Climate-driven declines in arthropod abundance restructure a rainforest food web. Proc Natl Acad Sci 115(44):E10397–E10406
Loboda S, Savage J, Buddle CM, Schmidt NM, Høye TT (2018) Declining diversity and abundance of High Arctic fly assemblages over two decades of rapid climate warming. Ecography 41(2):265–277
McKinney ML (1999) High rates of extinction and threat in poorly studied taxa. Conserv Biol 13(6):1273–1281
Meirmans PG, Van Tienderen PH (2004) genotype and genodive: two programs for the analysis of genetic diversity of asexual organisms. Mol Ecol Notes 4:792–794
Miller MA, Pfeiffer W, Schwartz T (2010) Creating the CIPRES Science Gateway for inference of large phylogenetic trees. Curran Associates, Red Hook, NY, pp 1–8
Mora C, Tittensor DP, Adl S, Simpson AG, Worm B (2011) How many species are there on earth and in the ocean? PLoS Biol 9(8):e1001127
Moritz C (1994) Defining ‘evolutionarily significant units’ for conservation. Trends Ecol Evol 9(10):373–375
Muñoz PT, Torres FP, Megías AG (2015) Effects of roads on insects: a review. Biodivers Conserv 24(3):659–682
Murphy SA, Joseph L, Burbidge AH, Austin J (2011) A cryptic and critically endangered species revealed by mitochondrial DNA analyses: the Western Ground Parrot. Conserv Genet 12(2):595–600
Paradis E (2010) pegas: an R package for population genetics with an integrated-modular approach. Bioinformatics 26:419–420
Peterson BK, Weber JN, Kay EH, Fisher HS, Hoekstra HE (2012) Double digest RADseq: an inexpensive method for de novo SNP discovery and genotyping in model and non-model species. PLoS ONE 7(5):e37135
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155(2):945–959
R Core Team (2015) R: a language and environment for statistical computing. Vienna, Austria: R foundation for statistical computing. https://www.R-project.org/
Régnier C, Fontaine B, Bouchet P (2009) Not knowing, not recording, not listing: numerous unnoticed mollusk extinctions. Conserv Biol 23(5):1214–1221
Régnier C, Achaz G, Lambert A, Cowie RH, Bouchet P, Fontaine B (2015) Mass extinction in poorly known taxa. Proc Natl Acad Sci 112(25):7761–7766
Rohland N, Reich D (2012) Cost-effective, high-throughput DNA sequencing libraries for multiplexed target capture. Genome Res 22(5):939–946
Rubinoff D (2001) Evaluating the California gnatcatcher as an umbrella species for conservation of coastal sage scrub. Conserv Biol 15:1374–1383
Rubinoff D (2006) Utility of mitochondrial DNA barcodes in species conservation. Conserv Biol 20(4):1026–1033
Rubinoff D, Sperling FAH (2004) Mitochondrial DNA sequence, morphology and ecology yield contrasting conservation implications for two threatened Buckmoths (Hemileuca: Saturniidae). Biol Conserv 118:341–351
Saitou N, Nei M (1987) The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol 4(4):406–425
Samways MJ (2007) Insect conservation: a synthetic management approach. Annu Rev Entomol 52:465–487
Sánchez-Bayo F, Wyckhuys KA (2019) Worldwide decline of the entomofauna: a review of its drivers. Biol Conserv 232:8–27
Shaffer H, Fellers GM, Randal Voss S, Oliver JC, Pauly GB (2004) Species boundaries, phylogeography and conservation genetics of the red-legged frog (Rana aurora/draytonii) complex. Mol Ecol 13(9):2667–2677
Simon C, Frati F, Beckenbach A, Crespi B, Liu H, Flook P (1994) Evolution, weighting, and phylogenetic utility of mitochondrial gene sequences and a compilation of conserved polymerase chain reaction primers. Ann Entomol Soc Am 87(6):651–701
Stamatakis A (2014) RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30(9):1312–1313. https://doi.org/10.1093/bioinformatics/btu033
Stein BA, Kutner LS, Hammerson GA, Master LL, Morse LE (2000) State of the states: geographic patterns of diversity, rarity, and endemism. Precious heritage: the status of biodiversity in the United States. Oxford University Press, New York, pp 119–158
Trontelj P, Fišer C (2009) Perspectives: cryptic species diversity should not be trivialised. Syst Biodivers 7(1):1–3
Vogel G (2017) Where have all the insects gone? Science 345(6338):576–579
United States Fish and Wildlife Services (2011) Determination of endangered status for Casey’s June beetle and designation of critical habitat; final rule. Fed Reg 76(184):58954–58998
United States Fish and Wildlife Services (2019) Listed species believed to or known to occur in each state. https://ecos.fws.gov/ecp0/reports/species-listed-by-state-totals-report. Accessed 15 Mar 2019
Winter DJ (2012) MMOD: an R library for the calculation of population differentiation statistics. Mol Ecol Res 12:1158–1160
Zhang J, Pei N, Mi X (2012) Phylotools: phylogenetic tools for Ecophylogenetics. R package version 0.1.2. https://CRAN.R-project.org/package=phylotools
Zheng X, Levine D, Shen J, Gogarten S, Laurie C, Wier B (2012) A high-performance computing toolset for relatedness and principal component analysis of SNP data. Bioinformatics 28(24):3326–3328. https://doi.org/10.1093/bioinformatics/bts606
Acknowledgements
We dedicate this paper to the memory of Chris Gregory, USFWS, who tirelessly advocated for the thorough investigation and protection of all threatened species, and without whom this project would have never been completed. Noelle Ronan (USFWS) provided valuable feedback, comments and support. ddRAD library construction was performed with the support of Angela Kauwe (USDA-ARS). HiSeq 4000 sequencing was provided by the Vincent J. Coates Genomics Sequencing Laboratory at UC Berkeley, supported by NIH S10 OD018174 Instrumentation Grant. This research was funded by the United States Fish and Wildlife Service, with additional funding provided by The College of Tropical Agriculture and Human Resources, University of Hawaii at Manoa; USDA Cooperative State Research, Education and Extension (CSREES), Grant/Award Number: HAW00942-H. USDA is an equal opportunity employer. Mention of trade names or commercial products in this publication is solely for the purpose of providing specific information and does not imply recommendation or endorsement by the USDA.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by Anurag Chaurasia.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The findings and conclusions in this article are those of the author(s) and do not necessarily represent the views of the U.S. Fish and Wildlife Service.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Rubinoff, D., Reil, J.B., Osborne, K.H. et al. Phylogenomics reveals conservation challenges and opportunities for cryptic endangered species in a rapidly disappearing desert ecosystem. Biodivers Conserv 29, 2185–2200 (2020). https://doi.org/10.1007/s10531-020-01968-w
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10531-020-01968-w