
Abstract
The South African scale insect Pulvinariella mesembryanthemi was introduced worldwide in several coastal areas with Mediterranean climate, probably through infested plants of Carpobrotus sp. Its high host specificity and its capacity to produce severe damages in the invasive Carpobrotus sp. plants makes this insect a potential biocontrol agent. To test the efficiency and host range of insects used for biocontrol, population genetic studies can help to unravel cryptic complexes and intraspecific diversity. In this study we performed a genetic analysis including native and exotic populations of P. mesembryanthemi, through Sanger sequencing of mitochondrial (cytochrome c oxidase I, COI) and ribosomal (D2–D3 expansion segments of the large subunit ribosomal RNA gene 28S) gene fragments. Accidentally, an endosymbiont was sequenced with one of the pair of primers used. The exotic populations of the insect did not show any variability among populations for both studied genes, which suggest a common origin of all studied introduced populations. Contrastingly, native populations showed high variability and seemed to be a cryptic species complex. Moreover, the Gauteng populations (from NE South Africa) were phylogenetically the closest to the exotic ones, suggesting that the exotic populations could be original from somewhere near this area. An endosymbiont of P. mesembryanthemi was detected, and the sequenced coxA gene was similar to that of the Rickettsiaceae family from the α-Proteobacteria, and close to other insect endosymbionts. To the best of our knowledge, this was the first mention of this endosymbiont in P. mesembryanthemi, although α-Proteobacteria endosymbionts have been reported for other sap-sucking insects.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The highly invasive plant Carpobrotus edulis (L.) N.E. Br. has colonized several temperate coastal regions around the world (D’Antonio et al. 1993; de la Peña et al. 2010; Delipetrou 2006), where it causes detrimental effects to other plant (Badalamenti et al. 2016; Jucker et al. 2013) and insect (Orgeas et al. 2007) species, negatively modifies community structure (Santoro et al. 2012) and can cause long-lasting changes to soil properties (Badalamenti et al. 2016; Molinari et al. 2007), delaying ecosystem restoration even after its removal (Conser and Connor 2009; Novoa et al. 2013). Currently, hand-removal and herbicides are the two main methods of control used (Delipetrou 2006); however, they are costly, time-consuming and usually ineffective, causing additional ecosystem damage (Carta et al. 2004; Fagúndez and Beiras 2007). As such, biological control presents itself as a potential option for the cost-effective control of C. edulis across its invaded range, although to date this has not yet been attempted for this invasive plant (DiTomaso and Kyser 2013). Biological control of C. edulis might be particularly effective given that Van Grunsven et al. (2009) found evidences that C. edulis is favored in invaded areas that do not have specific enemies.
The scale insect Pulvinariella mesembryanthemi (Vallot) (Family: Coccidae, Order: Hemiptera) has been observed feeding on C. edulis in its native and invasive ranges and presents itself as a potential biological control agent because of its high host specificity (Miller et al. 2005; Miller and Miller 2003) and its capability of producing severe damages to the plant (Donaldson et al. 1978; Washburn and Frankie 1985). This insect, as well as its host plant, originates from South Africa (Washburn and Frankie 1985), from where it may have been accidentally introduced in several parts of the world along with infested plants. Currently, it is present in Southern Europe (Gómez-Menor Ortega 1954; Kozar et al. 2016; Pellizzari and Germain 2010; Seljak 2010; Vieira et al. 1983), Turkey (Cebeci and Selmi 2004), Argentina (Granara de Willink and Claps 2003), USA (Miller et al. 2005), New Zealand (Hodgson and Henderson 2000) and Australia (Collins and Scott 1982).
However, cryptic species complexes (those species classified as a single species by their morphology; Darlington 1940) are common in scale insects, but frequently overlooked by morphological identification (Andersen et al. 2010a). As a result, genetic studies of potential biological control agents (not only scale insects) are highly recommended in order to uncover cryptic species complexes or intraspecific entities prior to release or promotion, which cannot be distinguished with morphological characteristics (Gaskin et al. 2011; Paterson et al. 2016; Rosen 1986).The presence of cryptic species can affect control success by having individuals that show different host specificity or aggressiveness towards the plant (Rauth et al. 2011) and/or that respond differently to the climatic conditions (Paterson et al. 2016) present among the released/promoted agents. This is especially important when using allopatric populations as biocontrol agents. In the case of P. mesembryanthemi, the semi-cryptic species Pulvinaria delottoi Gill has been cited in California (Washburn and Frankie 1985) and the UK (Salisbury et al. 2011). They are almost morphologically identical, but with different feeding habits and life cycle duration: P. mesembryanthemi prefers younger leaves and has two reproductive events per year, while P. delottoi usually feeds on older leaves and has one generation per year in Southern California (Washburn and Frankie 1981, 1985). Their reproduction seems to be exclusively parthenogenic, being able of producing up to 2400 hatches per ovisac, with an average fecundity of 350–800 crawlers per individual and higher for P. delottoi than for P. mesembryanthemi (Washburn and Frankie 1985). Although P. mesembryanthemi males have been observed, no sexual reproduction has been recorded and the presence of male individuals has been related to a relictic species feature (Washburn and Frankie 1985).
Genetic studies are also particularly important for studies of scale insects, as well as other sap-sucking Hemiptera, as these species have both obligate and facultative associations with bacterial endosymbionts (Moran et al. 2008), though for scale insects these are still beginning to be studied (Gruwell et al. 2007). Many of these endosymbionts are vertically transmitted (from the mother to the progeny) (Bing et al. 2013; Li et al. 2011b), making themselves useful for phylogeny reconstructions (Andersen et al. 2010b) and insect identification (Mathenge et al. 2015). Others may be horizontally transmitted, such as the reproductive manipulator Wolbachia that can also alter the diversity of the frequency of its host’s mitochondrial DNA haplotypes in a population (Mathenge et al. 2015). However, it was also reported that not all endosymbionts affect host phenotype. For example, according to Hamm et al. (2014), Wolbachia had not detectable effects on their host. As such, identifying the presence of microbial symbionts can be important both for species and population-level identifications and for understanding potential reproduction-disrupting effects for insects considered for biological control programs (Mathenge et al. 2015). However, to the extent of our knowledge, no studies have been conducted to examine the population structure of P. mesembryanthemi or to examine the existence of its endosymbionts.
For insects, the mitochondrial gene cytochrome c oxidase I (COI) is frequently used to compare interspecific genetic diversity and to discover cryptic biodiversity (Bekker et al. 2016; Mesquita Fonseca et al. 2017; Porco et al. 2012). While the “DNA barcode region” of COI (Hebert et al. 2003) is widely used for species delimitation and identification (Wang et al. 2015), recent studies have found that the standard set of primers used for insect COI sequencing have failed for some species of scale insects, and some authors recommend the nuclear large subunit ribosomal RNA gene (LSU rRNA, 28S) to be used as a substitute in this group (Schroer et al. 2008), or a concatenation of the 28S and COI gene fragments (Sethusa et al. 2014). Interestingly, for many insect groups 28S sequences are more conservative than those of COI (Wang et al. 2015); however, Schroer et al. (2008) found that 28S could be more variable than COI in some scale insects.
Therefore, the aim of this study was to analyse both the intra- and inter- population genetic variability of the insect P. mesembryanthemi by sequencing fragments of COI and 28S from individuals in its native and non-native populations with the goal of providing genetic resources that may be helpful for future studies of the use of this species as a biocontrol agent for C. edulis. In addition, we seek to explore the origins of populations of P. mesembryanthemi that have been introduced globally, and to examine the presence of microbial endosymbionts.
Materials and methods
Sample collection and preservation
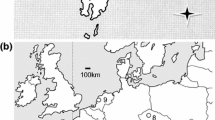
Samples were taken from 18 populations worldwide (12 from the Iberian Peninsula, 2 from the Canary Islands, 1 from New Zealand and 3 from South Africa) (for collection information, see Fig. 1 and Online Resource 1) during 2016–2017. The samples were taken from females preferentially in the 4th instar as described by Washburn and Frankie (1985). Careful visual inspection of the samples was done to detect parasitized individuals, which were discarded. Specimens were preserved in 70% to 96% ethanol and stored at − 20 °C.

Origin of populations of Pulvinariella mesembryanthemi used in this study. SPAIN: 1—Santander (STD), 2—Asturias (AST), 3—Oleiros (MER), 4—A Coruña (CNH), 5—Punta Nariga (NAR), 6—Sálvora island (SAL), 7—Cádiz (CAD), 8—Tenerife island (TEN), 9—La Palma island (LPA). PORTUGAL: 10—Lisboa (LTE), 11—Cascais (CCS), 12—Estoril (EST), 13—Costa de Caparica (CAP). NEW ZEALAND: 14—North New Brighton (NZL). AUSTRALIA: 15—New South Wales (AUS). SOUTH AFRICA: 16,18,19—Gauteng (SA1, SA3, SAP), 17—Eastern Cape (SA2)
DNA extraction, amplification and sequencing
DNA was extracted from whole individuals with the E.Z.N.A. ® Tissue DNA kit (Omega bio-tek) and the mitochondrial locus COI and the ribosomal gene 28S were amplified as follows. For COI, initially the primers LepF and LepR (Hajibabaei et al. 2006) were used; however, a search of the resulting sequences with BLAST in NCBI (http://www.ncbi.nih.gov/BLAST/) showed that the obtained COI sequence (633 bp) belonged to the α-Proteobacteria secondary endosymbiont of the aphid Sitobian miscanthi (HQ645970; Li et al. 2011a, b). We then utilized the primer-pair of PcoF1 and LepR and the protocol presented in Park et al. (2010) and the resulting sequences (679 bp) were confirmed to be insect COI based on BLAST searches. For 28S, the D2 and D3 expansion segments of the nuclear large subunit ribosomal RNA gene (LSU rRNA, 28S) (fragment of 820 bp) were amplified with the pair of primers s3660/A335 and the protocol presented in Schroer et al. (2008). PCRs were performed in a total volume of 20 μL comprising 2.5 μL (5.0 μL when amplifying the endosymbionts) of nucleic acid solution (10–35 ng/μL) and 17.5 μL of mix (4 μL of 5 × Buffer, 25 mM MgCl2, 2.5 mM dNTPs, 8 pmol of each primer and 0.8 U of GoTaq DNA Polimerase) on a Biorad thermocycler and PCR products were visualized on 0.5% agarose gel, before being purified with SureClean Kit and sent to Genewiz Inc. sequencing.
The COI fragment of P. mesembryanthemi was sequenced for one individual of each location, except for the South African populations where 8 (for SA2 and SA3) or 7 (for SA1) individuals were sequenced. For the 28S fragment of P. mesembryanthemi, one individual of the populations NZL, AST, SA1, SA3, SA2 (see Online Resource 1) were sequenced. The coxA fragment of the endosymbiont was sequenced for one individual of the populations AST, NAR, TEN, CCS, AUS and SA1 (see Online Resource 1). However, the SA2 population could not be included for the endosymbiont analysis, as a parasitoid of the insect was sequenced instead. When that sequence was searched in the BLAST tool of the NCBI (https://blast.ncbi.nlm.nih.gov/), a match of 84% similarity with two genera of the Chalcidoidea parasitic wasps (Encarsia sp. and Encyrtidae sp.) was found. The identification of a Chalcidoidea wasp in one of our P. mesembryanthemi populations is not surprising, as it is quite common to recover DNA from this parasitoid from scale insect studies (Gwiazdowski and Normark 2014).
Phylogenetic analysis
Sequences of the insect and its endosymbiont obtained from BOLD System (www.boldsystem.org) were included in the analysis (see Online Resource 1 for accession numbers). The COI sequences of P. mesembryanthemi from Chile which are available in the NCBI (accession numbers: KY085884-KY085888, KY085331-KY085334) were not included in the analysis as they seemed to be misidentified – the plants from which they were sampled do not match its suitable host plants (and neither those of P. delottoi) (Washburn and Frankie 1985) and their similarity with other COI sequences of P. mesembryanthemi obtained in this study and from the BOLD System are quite low (16–18% uncorrected p-distances).
After checking sequences identity with the BLAST tool from the NCBI website, forward and reverse sequences were assembled and edited with Sequencher 4.0.5 (Sequencher®) and then aligned with ClustalW Multiple Aligment function (Thompson et al. 1994) through BioEdit v.7.0.5.2 (Hall 1999). For the analysis, only unique haplotypes were included.
Analyses were done with the COI and 28S gene fragments of P. mesembryanthemi both individually and concatenated [using Concatenator v 1.0.1. (Pina-Martins and Paulo 2008) to combine the sequences]. For the endosymbiont, analyses were only done with the coxA gene fragment. The most suitable models of DNA substitutions to the data for each dataset were found with jModelTest 2.1.10 (Darriba et al. 2012; Guindon and Gascuel 2003). Those models were then used to infer the phylogenetic trees by Maximum Likelihood (ML) with PhyML 3.0 (Guindon et al. 2010), and by Bayesian Inference (BI) with MrBayes 3.2 (Huelsenbeck and Ronquist 2001). For the BI analysis with the concatenated dataset we used for each partition (COI, 28S gene fragments) the best-fit model corresponding to the COI or 28S data, respectively. Phylogenetic trees were also inferred by Maximum Parsimony (MP) with PAUP* 4.0a152 (L. Swofford 2002). MP analysis were performed with a heuristic search with TBR (tree bisection reconnection) branch swapping of 100 random sequence addition starting trees, and branch support was obtained from 1000 pseudo-replicates of nonparametric bootstrap. ML analysis were performed with SPR (sub-tree pruning and re-grafting) branch swapping of 10 random starting trees, and branch support was obtained from 1000 bootstrap pseudo-replicates. For the BI, posterior probabilities were estimated by a Metropolis-coupled Markov chain Monte Carlo sampling algorithm, and 2,000,000 generations were sampled every 1500 generations. Stationarity of the likelihood scores with the generation time was checked with TRACER v1.6 (Rambaut et al. 2018). Nine other Coccidae species and one Diaspididae species were used as outgroups in the P. mesembryanthemi trees (see Online Resource 2 for accession numbers). For the endosymbionts trees, five other species (five of them Proteobacteria, one Heterokontophyla and another Ochrophyta) were used (see Online Resource 3 for accession numbers).
Pairwise divergences were calculated through the most suitable model for each dataset (as found with jModelTest) and the Kimura two-parameter (K2P) model with PAUP*. The K2P distances were included for comparison with other studies. Descriptive parameters such as haplotype and nucleotide diversity were calculated with DNASP 5.10 (Librado and Rozas 2009).
Results
Pulvinariella mesembryanthemi populations: COI gene
The obtained sequences had an average GC composition of 25% (Table 1). No gaps were found in the COI fragments. Haplotype diversity for the COI gene was high in the native area (Hd = 0.757), where 5 different haplotypes were found, most likely corresponding to different species, whilst only one haplotype was found in the introduced area (EXT) (Fig. 2; Online Resource 4). In the native area, there were 129 polymorphic sites, all of them parsimony informative; and 141 mutations (Online Resource 4). The only native population where intra-population variability was found (of the three native populations in which 7–8 samples were studied) was SA1, with two haplotypes (SA1.1, SA1.2).

Maximum likelihood tree based on COI data of scale insects. Samples from Pulvinariella mesembryanthemi have been marked in bold and the name of the haplotype is presented (see Online resource 1). The tree was rooted with Aspidiotus nerii. On the right side of the figure, the families of the scale insects are indicated. The numbers next to the nodes represent, in this order, the bootstrap support values (≥ 50%) of maximum likelihood and maximum parsimony. Numbers within parenthesis represent the Bayesian posterior probability (≥ 0.5). See Online resource 2 for NCBI codes
The distances calculations showed that the sole exotic haplotype found for P. mesembryanthemi was most similar to two South African populations from Pretoria (SAP, SA3), having a 3 and 5% of TrN model and K2P distances from them for this gene fragment (Tables 1, 2). The most divergent South African populations from the exotic ones were also from Pretoria (SA1.1, SA1.2), and had a 12–15% of TrN distance (15% K2P distance) (Table 2). The South African population from Eastern Cape Province (SA2) had an intermediate TrN distance of 11% (13% K2P distance) (Table 2). Also, the Pretoria populations SA1 and SA3 had a 14–15% genetic TrN distance (17% K2P distance), slightly higher to the distance between the Pretorian SA1 and the one from Eastern Cape Province SA2 (10% TrN and 12–13% K2P distances) (Table 2). The highest similarities between P. mesembryanthemi haplotypes corresponded to the pairs of haplotypes SA3 - SAP (TrN and K2P distance of 0.1 and 0.2%) and SA1.1–SA1.2 (TrN and K2P distance of 0.2–0.3%) (Table 2).
Regarding the topology of the trees for the P. mesembryanthemi samples, it was quite similar for all methods (Fig. 2, Online Resource 5, Online Resource 6). All the haplotypes were grouped in a clade with a high support from BI posterior probabilities, but with low support from ML and MP bootstrap values (Fig. 2). A clade formed by the exotic populations and the Pretorian populations SA3 and SAP was highly supported by both bootstrap values and by the BI posterior probability (Fig. 2). The three trees grouped the haplotypes SA1.1 and SA1.2 with SA2, although with a low bootstrap value (Fig. 2).
Pulvinariella mesembryanthemi populations: 28S gene
An average GC composition of 61% was found in the sequences (Online Resource 4). The 28S sequenced region did not show variability between exotic population, but it distinguished two genotypes in the South African populations (SA1, SA2) which were different from the exotic one (Fig. 3; Online Resource 4). There were 18 polymorphic sites, only two of them parsimony informative (Online Resource 2). Native populations had a haplotype diversity of 0.67 and 16 mutations (Online Resource 4). From the analysed sequences of P. mesembryanthemi only one South African population (SA2) showed indels (Online Resource 4).

Maximum likelihood tree based on 28S data of scale insects. Samples from Pulvinariella mesembryanthemi have been marked in bold and the name of the haplotype is presented (see Online resource 1). The tree was rooted with Aspidiotus nerii. On the right side of the figure, the families of the scale insects are indicated. The numbers next to the nodes represent, in this order, the bootstrap support values (≥ 50%) of maximum likelihood and maximum parsimony. Numbers within parenthesis represent the Bayesian posterior probability (≥ 0.5). See Online resource 2 for NCBI codes
From the distances calculations, the most similar populations to the exotics were the South African ones from Pretoria (SA1, SA3; with 0.2% HKY model and K2P distances), whilst the most different one was the South African population from Eastern Cape Province (2.2% distance with both methods) (Tables 1, 2). The distance between the Pretorian genotype (SA1) and SA2 was of 2.0% (with both methods) (Table 2).
Trees estimated by Maximum Likelihood (Fig. 3), Maximum Parsimony (Online Resource 7) and Bayesian Inference (Online Resource 8) included a monophyletic assemblage for the P. mesembryanthemi haplotypes, which was highly supported by bootstrap and posterior probability (Fig. 3). The P. mesembryanthemi exotic genotype (EXT) was nested with the native haplotype SA1 in the MP and BI trees, although with low support (Online Resource 7, Online Resource 8). However, the ML tree nested the two native haplotypes (SA1, SA2) with a low support (Fig. 3).
Pulvinariella mesembryanthemi populations: COI + 28S genes
The South African population SA3 from Pretoria was the native populations that showed a lower genetic distance to the exotic ones, and therefore the most similar one (Table 2).
As in the 28S trees, P. mesembryanthemi was recovered as a clade with a strong support of all methods (Fig. 4). The topology for the P. mesembryanthemi haplotypes varied between methods (Fig. 4, Online Resource 9, Online Resource 10). Independent of the method, there was a high support for a clade with the exotic haplotype (EXT) and one of the Pretorian haplotypes (SA3) (Fig. 4). There was a high support of ML bootstrap value and BI posterior probability for a clade between these two haplotypes (EXT, SA3) and the other Pretorian haplotype (SA1) (Fig. 4). However, the MP tree nested SA1 and SA2 haplotypes with a very low support (< 50%) (Online Resource 9).

Maximum likelihood tree based on the concatenation of COI and 28S data of scale insects. Samples from Pulvinariella mesembryanthemi have been marked in bold and the name of the haplotype is presented (see Online resource 1). The tree was rooted with Aspidiotus nerii. On the right side of the figure, the families of the scale insects are indicated. The numbers next to the nodes represent, in this order, the bootstrap support values (≥ 50%) of maximum likelihood and maximum parsimony. Numbers within parenthesis represent the Bayesian posterior probability (≥ 0.5). See Online resource 2 for NCBI codes
Endosymbiont: coxA gene
The amplification of the coxA region of this endosymbiont did not showed any variability between exotic and native populations, except for the Australian population, which slightly differed from the others due to one single mutation.
The two endosymbiont’s haplotypes that were found only differed a 0.3% model corrected distance (Tables 1, 3). This P. mesembryanthemi endosymbiont was related the most to S. mischanthi endosymbiont (6% GTR distance), and then to Wolbachia sp. and Orientia tsutsugamushi (Table 3).
The two haplotypes of P. mesembryanthemi endosymbiont were grouped with a high support of the three tested methods (Fig. 5, Online Resource 11, Online Resource 12). The three Hemiptera endosymbiont haplotypes (two from P. mesembryanthemi and one from S. miscanthi) were nested with a high support of both bootstrap values and posterior probability (Fig. 5). The maximum likelihood tree was the only one to group the Hemiptera endosymbionts with Wolbachia sp. (an Arthropoda endosymbiont), although with a very low support (53%) (Fig. 5). With all methods, all the endosymbionts included in the analysis were grouped with a very high support (Fig. 5).

Maximum likelihood tree based on coxA data of the Pulvinariella mesembryanthemi endosymbiont and its closest taxa. Samples from the P. mesembryanthemi endosymbiont have been marked in bold and the name of the haplotype is presented (see Online resource 1). The tree was rooted with Kuckuckia spinosa. On the left side of the figure, the class and phylum of the taxa are indicated, whereas in the right side the host species and their order and class are showed. The numbers next to the nodes represent, in this order, the bootstrap support values (≥ 50%) of maximum likelihood and maximum parsimony. Numbers within parenthesis represent the Bayesian posterior probability (≥ 0.5). See Online resource 3 for NCBI codes
Discussion
Pulvinariella mesembryanthemi populations
Pairwise distances between the studied populations were quite high in some cases, suggesting that P. mesembryanthemi is a cryptic species complex. Amouroux et al. (2017) delimited the intra-specific K2P distance for the COI 5′ gene fragment to 0–2% and for the 28S gene fragment to 0–0.6% for Coccidae. Only the haplotype pairs SA3-SAP and SA1.1-SA1.2 (for COI) and SA1-EXT (for 28S) lay within these intervals. Amouroux et al. (2017) also found a gap region (from 2.0 to 12.5% K2P distance) for their Coccidae samples between the intra-specific and inter-specific distances. However, in our COI samples the K2P distance between the exotic haplotype and SA3 or SAP is of 5%. The K2P distances between SA1 or SA2 and the rest of haplotypes would lie in the inter-specific interval found by Amouroux et al. (2017) for the 5′ COI region. Distances between SA2 and SA1 or EXT would also lie within the inter-specific interval found in Amouroux et al. (2017) for the 28S gene fragment. Another study on Coccidae found a wider intra-specific interval (0–4.2% K2P distance for COI and 0–1.25% for 28S), but a much narrower gap (4.2–4.6% for COI) between intra-specific and inter-specific distances (Wang et al. 2015). The intra-specific and inter-specific intervals of the COI barcode region are usually well separated (Hebert et al. 2003), but in some cases they can overlap (Davison et al. 2009).
The exotic populations of P. mesembryanthemi included in the analysis did not show any variability between populations for the two sequenced gene fragments, even when comparing populations from Europe and Oceania. The absence of genetic variability for the studied genes suggest that there was one single genotypic and geographic origin from South Africa of the different exotic populations of the insect, from where it expanded to the different areas, and that they did not diverged since then. However, these results could also be due to a lack of resolution of the selected loci to determine if differentiation between the exotic populations has occurred.
This lack of genetic variability in the introduced range has also been seen in studies of other scale insects (Schroer et al. 2008). Also, if we take into account that P. mesembryanthemi has only parthenogenetic reproduction (Nur 1963; Pesson 1941), even a single female might have been the origin of the exotic populations. As this insect has very high host specificity (Washburn and Frankie 1985), it is probable that the same Carpobrotus sp. plants that hosted the original insect population were dispersed to the different exotic localities included in this study. Another possibility is that Carpobrotus sp. plants were exported multiple times from the same source location. Either way, loci with higher resolution will be necessary to confirm that all exotic populations come from a unique one in the native area. In addition, the parthenogenetic reproduction together with the fairly recent introduction outside the native range [the first records we found of the insect in Europe and Oceania are from the 1980s (Collins and Scott 1982; Vieira et al. 1983)] might have contributed to the genetic homogeneity of the species in the introduced area.
The origin of the exotic populations of P. mesembryanthemi seems to be near Pretoria, since the native SA3 was the most similar to them. However, a wider sampling in South Africa would be necessary to determine the precise origin of the introduced insects. Apparently, none of the native populations we included in this study was the origin of the exotic ones, and maybe none of them was the same species as the exotic, given that the genetic distances between them were quite high for the COI gene fragment (> 5% K2P distance). However, a finer scale analysis and morphological analysis would be needed to determine if the EXT populations and their closer native populations SA3 and SAP are indeed different species.
Contrarily to what happened with P. mesembryanthemi exotic populations, native populations showed between samples variability and even, for one of the populations, slightly different haplotypes within it. This high variability between native populations (most of them showed > 10% K2P distance for the COI gene fragment) indicates that they are a cryptic species complex. This could also explain why a slightly higher genetic distance was found between SA1 and SA3 populations (both from Pretoria and separated by approximately 12 km) than between SA1 and SA2 populations (separated roughly 900 km) for the COI gene fragment.
As much higher genetic diversity was found in native areas compared to exotic areas of the insect, optimization experiments for the use of P. mesembryanthemi as a biocontrol agent should test native populations: that is, where most of the genetic diversity can be found, and consequently, more chances of finding differences on host range, climate adaptability or capability of damaging the plant would be found.
Endosymbiont
Symbiosis of Hemiptera insects with α-Proteobacteria have been documented for several other species (Bing et al. 2013; Brady et al. 2014; Li et al. 2011b; Mathenge et al. 2015). In this study, the presence of an α-Proteobacteria endosymbiont in P. mesembryanthemi was confirmed by the sequences obtained with the pair of primers LepF/LepR. The closest specimen found in the NCBI database was the endosymbiont SMLS (Sytobian miscanthi L type symbiont), which is as α-Proteobacteria from the Rickettsiaceae family (Li et al. 2011a, b). They were very closely related, with a similarity for the coxA fragment of 95%. SMLS was found to give some fitness advantages (Li et al. 2016) and to be widely present in the populations of its host insect (Li et al. 2011b), which belongs to the Hemiptera order as P. mesembryanthemi. In our study, endosymbiont genetic material was amplified from all the populations we tried (5 of them exotic and 2 native), with the exception of the SA2 population (although this could be related to a low specificity of the primers or of the extraction technique, as a parasitoid was sequenced in its place).
The sequenced endosymbionts of the exotic populations were identical for the chosen gene fragment to the native ones, with the exception of one exotic population that had a small difference. The variability of the endosymbiont was therefore much more reduced than the variability found for P. mesembryanthemi. These results are in accordance to the low genetic diversity between populations that Li et al. (2011b) also found for SMLS, and which they related to a recent and frequent intraspecific transfer. The α-Proteobacteria endosymbionts found in Hemiptera insects are closely related to Rickettsiaceae bacteria (Bing et al. 2013; Brady et al. 2014; Mathenge et al. 2015). For instance, Li et al. (2011a) found a very close relationship between SMLS and the Rickettsiaceae bacteria Orientia tsutsugamushi (94% of similarity for the 16S sequence, although probably from different genera), whilst in our case the similarity between our endosymbiont and a coxA sequence from O. tsutsugamushi (GBBAC698-15) was of 75%.
Conclusions
Ours results, which distinguish only one haplotype for all the exotic P. mesembryanhtemi populations in both genes, suggest a common origin of all of them, which seems not to be any of the native ones sampled for this study. Most of the genetic diversity found for this insect resided in its native range, South Africa, suggesting that P. mesembryanthemi is a cryptic species complex. Therefore, when targeting the insect P. mesembryanthemi as a biological control of the invasive C. edulis, studies on the optimization of the control should preferentially compare different South African populations, where more variability would be found.
In addition, this study recovered α-Proteobacteria sequences from the insect samples, which were very close to those of other Hemiptera endosymbionts. To the extent of our knowledge, this was the first mention of an endosymbiont in P. mesembryanthemi. The analysed sequences of this P. mesembryanthemi endosymbiont were almost identical, even when comparing exotic and native populations.
References
Amouroux P et al (2017) Genetic diversity of armored scales (Hemiptera: Diaspididae) and soft scales (Hemiptera: Coccidae) in Chile Sci Rep-UK 7:2014 https://doi.org/10.1038/s41598-017-01997-6
Andersen JC, Gruwell ME, Morse GE, Normark BB (2010a) Cryptic diversity in the Aspidiotus nerii complex in Australia. Ann Entomol Soc Am 103:844–854. https://doi.org/10.1603/AN10060
Andersen JC, Wu J, Gruwell ME, Gwiazdowski R, Santana SE, Feliciano NM, Morse GE, Normark BB (2010b) A phylogenetic analysis of armored scale insects (Hemiptera: Diaspididae), based upon nuclear, mitochondrial, and endosymbiont gene sequences. Mol Phylogenet Evol 57(3):992–1003
Badalamenti E, Gristina L, Laudicina VA, Novara A, Pasta S, La Mantia T (2016) The impact of Carpobrotus cfr. acinaciformis (L.) L. Bolus on soil nutrients, microbial communities structure and native plant communities in Mediterranean ecosystems. Plant Soil 409:19–34. https://doi.org/10.1007/s11104-016-2924-z
Bekker EI, Karabanov DP, Galimov YR, Kotov AA (2016) DNA barcoding reveals high cryptic diversity in the North Eurasian Moina species (Crustacea: Cladocera). PLoS ONE 11:e0161737. https://doi.org/10.1371/journal.pone.0161737
Bing X-L, Yang J, Zchori-Fein E, Wang X-W, Liu S-S (2013) Characterization of a newly discovered symbiont of the whitefly Bemisia tabaci (Hemiptera: Aleyrodidae). Appl Environ Microb 79:569–575. https://doi.org/10.1128/aem.03030-12
Brady CM et al (2014) Worldwide populations of the aphid Aphis craccivora are Infected with diverse facultative bacterial symbionts. Microb Ecol 67:195–204. https://doi.org/10.1007/s00248-013-0314-0
Carta L, Manca M, Brundu G (2004) Removal of Carpobrotus acinaciformis (L.) L. Bolus from environmental sensitive areas in Sardinia, Italy. Paper presented at the proceedings 10th MEDECOS conference, Rhodes, Greece, 25/04/2004
Cebeci H, Selmi E (2004) The Coccidae species of Turkey. İstanbul Üniversitesi Orman Fakültesi Dergisi 54:207–228
Collins L, Scott J (1982) Interaction of ants, predators and the scale insect, Pulvinariella mesembryanthemi, on Carpobrotus edulis, an exotic plant naturalized in Western Australia. Aust Ent Mag 8:73–78
Conser C, Connor E (2009) Assessing the residual effects of Carpobrotus edulis invasion, implications for restoration. Biol Invasions 11:349–358. https://doi.org/10.1007/s10530-008-9252-z
Cook LG, Rowell DM (2007) Genetic diversity, host-specificity and unusual phylogeography of a cryptic, host-associated species complex of gall-inducing scale insects. Ecol Entomol 32:506–515. https://doi.org/10.1111/j.1365-2311.2007.00893.x
D’Antonio CM, Odion DC, Tyler CM (1993) Invasion of Maritime Chaparral by the Introduced Succulent Carpobrotus edulis. Oecologia 95:14–21
Darlington CD (1940) Taxonomic systems and genetic systems. In: Huxley J (ed) The new systematics. Clarendon Press, Oxford, pp 137–160
Darriba D, Taboada GL, Doallo R, Posada D (2012) jModelTest 2: more models, new heuristics and parallel computing. Nat Meth 9:772–772. http://www.nature.com/nmeth/journal/v9/n8/abs/nmeth.2109.html#supplementary-information
Davison A, Blackie RL, Scothern GP (2009) DNA barcoding of stylommatophoran land snails: a test of existing sequences. Mol Ecol Resour 9:1092–1101. https://doi.org/10.1111/j.1755-0998.2009.02559.x
de la Peña E, de Clercq N, Bonte D, Roiloa S, Rodríguez-Echeverría S, Freitas H (2010) Plant-soil feedback as a mechanism of invasion by Carpobrotus edulis. Biol Invasions 12:3637–3648. https://doi.org/10.1007/s10530-010-9756-1
Delipetrou P (2006) Carpobrotus edulis. http://www.europe-aliens.org/pdf/Carpobrotus_edulis.pdf. Accessed 21/11/2016 2016
DiTomaso JM, Kyser GB (2013) Iceplant (Hottentot fig). In: Weed control in natural areas in the western United States. University of California Weed Reseach and Information Center
Donaldson DR, Moore WS, Koehler CS, Joos JL (1978) Scales threaten iceplant in Bay Area. Calif Agric 32:4–7
Fagúndez J, Beiras MB (2007) Plantas invasoras de Galicia: bioloxía, distribución e métodos de control. Dirección Xeral de Conservación da Natureza, Santiago de Compostela
Gaskin JF et al (2011) Applying molecular-based approaches to classical biological control of weeds. Biol Control 58:1–21. https://doi.org/10.1016/j.biocontrol.2011.03.015
Gómez-Menor Ortega J (1954) Adiciones a los Cóccidos de España (tercera nota). CSIC - Instituto Español de Entomología, Consejo Superior de Investigaciones Científicas (España)
Granara de Willink M, Claps L (2003) Cochinillas (Hemiptera: Coccoidea) presentes en plantas ornamentales de la Argentina. Neotrop Entomol 32:625–637
Gruwell ME, Morse GE, Normark BB (2007) Phylogenetic congruence of armored scale insects (Hemiptera: Diaspididae) and their primary endosymbionts from the phylum Bacteroidetes. Mol Phylogenet Evol 44:267–280. https://doi.org/10.1016/j.ympev.2007.01.014
Guindon S, Gascuel O (2003) A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol 52:696–704
Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W, Gascuel O (2010) New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol 59:307–321
Gwiazdowski RA, Normark BB (2014) An unidentified parasitoid community (Chalcidoidea) is associated with pine-feeding Chionaspis Scale Insects (Hemiptera: Diaspididae). Ann Entomol Soc Am 107(2):356–363
Hajibabaei M, Janzen DH, Burns JM, Hallwachs W, Hebert PD (2006) DNA barcodes distinguish species of tropical Lepidoptera. Proc Natl Acad Sci USA 103:968–971. https://doi.org/10.1073/pnas.0510466103
Hall TA (1999) BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp Ser 41:95–98
Hamm CA, Begun DJ, Vo A, Smith CC, Saelao P, Shaver AC, Jaenike J, Turelli M (2014) Wolbachia do not live by reproductive manipulation alone: infection polymorphism in Drosophia suzukii and subpulchrella. Mol Ecol 23(19):4871–4885
Hebert PDN, Cywinska A, Ball SL, deWaard JR (2003) Biological identifications through DNA barcodes. Proc. R. Soc. Lond. B 270:313–321. https://doi.org/10.1098/rspb.2002.221
Hodgson CJ, Henderson RC (2000) Coccidae (Insecta: Hemiptera: Coccoidea). Fauna N Z 41:1–264
Huelsenbeck JP, Ronquist F (2001) MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics 17:754–755
Jucker T, Carboni M, Acosta ATR (2013) Going beyond taxonomic diversity: deconstructing biodiversity patterns reveals the true cost of iceplant invasion. Divers Distrib 19:1566–1577. https://doi.org/10.1111/ddi.12124
Kozar F, Paloukis S, Papadopoulos N (2016) New scale insects (Homoptera: Coccoidea) in the Greek entomofauna. Entomol Hell 9:63–68
Lázaro-Ibiza B (1900) Contribuciones a la flora de la Península Ibérica. Notas críticas acerca de la flora española Anales de la Sociedad Española de Historia Natural 29:125–176
Li T, Xiao J-H, Xu Z-H, Murphy RW, Huang D-W (2011a) Cellular tropism, population dynamics, host range and taxonomic status of an aphid secondary symbiont, SMLS (Sitobion miscanthi L Type Symbiont). PLoS ONE 6:21944. https://doi.org/10.1371/journal.pone.0021944
Li T, Xiao J-H, Xu Z-H, Murphy RW, Huang D-W (2011b) A possibly new Rickettsia-like genus symbiont is found in Chinese wheat pest aphid, Sitobion miscanthi (Hemiptera: Aphididae). J Invertebr Pathol 106:418–421. https://doi.org/10.1016/j.jip.2010.12.003
Li T et al (2016) The genetic diversity of SMLS (Sitobion miscanthi L type symbiont) and its effect on the fitness, mitochondrial DNA diversity and Buchnera aphidicola dynamic of wheat aphid, Sitobion miscanthi (Hemiptera: Aphididae). Mol Ecol 25:3142–3151. https://doi.org/10.1111/mec.13669
Librado P, Rozas J (2009) DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25:1451–1452. https://doi.org/10.1093/bioinformatics/btp187
Madeira PT, Tipping PW, Gandolfo DE, Center TD, Van TK, O’Brien WO (2006) Molecular and morphological examination of Cyrtobagous sp. collected from Argentina, Paraguay, Brazil, Australia, and Florida. Biol Control 51:679–701
Mathenge CW, Riegler M, Beattie GA, Spooner-Hart RN, Holford P (2015) Genetic variation amongst biotypes of Dactylopius tomentosus. Insect Sci 22:360–374. https://doi.org/10.1111/1744-7917.12120
Mesquita Fonseca P, Silva Loreto EL, Silva Gottschalk M, Jaqueline Robe L (2017) Cryptic diversity and speciation in the Zygothrica genus group (Diptera, Drosophilidae): the case of Z. vittimaculosa Wiedemann. Insect Syst Evolut 48:285–313
Miller GL, Miller DR (2003) Invasive soft scales (Hemiptera: Coccidae) and their threat to U.S. agriculture. Proc Entomol Soc Wash 105:832–846
Miller DR, Miller G, Hodges GS, Davidson JA (2005) Introduced scale insects (Hemiptera: Coccoidea) of the United States and their impact on U.S. agriculture. Proc Entomol Soc Wash 107:123–158
Molinari N, D’Antonio C, Thomson G (2007) 7 - Carpobrotus as a Case Study of the Complexities of Species Impacts. In: Cuddington K, Byers JE, Wilson WG, Hastings A (eds) Theoretical Ecology Series, vol 4. Academic Press, New York, pp 139–162. https://doi.org/10.1016/s1875-306x(07)80009-8
Moran NA, McCutcheon JP, Nakabachi A (2008) Genomics and evolution of heritable bacterial symbionts. Annu Rev Genet 42:165–190
New-Zealand-Plant-Conservation-Network (2014) Carpobrotus edulis. http://www.nzpcn.org.nz/flora_details.aspx?ID=3638. Accessed 03/08/2017 2017
Novoa A, Gonzalez L, Moravcova L, Pysek P (2013) Constraints to native plant species establishment in coastal dune communities invaded by Carpobrotus edulis: Implications for restoration. Biol Conserv 164:1–9
Nur U (1963) Meiotic parthenogenesis and heterochromatization in a soft scale, Pulvinaria hydrangeae (Coccoidea: Homoptera). Chromosoma 14:123–139. https://doi.org/10.1007/bf00336754
Orgeas J, Ponel P, Fadda S, Matock A, Turpaud A (2007) Conséquences écologiques de l’envahissement des griffes de sorcière (Carpobrotus spp.) sur les communautés d’insectes d’un îlot du Parc national de Port-Cros (Var) Scientific reports of Port-Cros national park 22:233-257
Park D-S, Suh S-J, Oh H-W, Hebert PD (2010) Recovery of the mitochondrial COI barcode region in diverse Hexapoda through tRNA-based primers. BMC Genom 11:423. https://doi.org/10.1186/1471-2164-11-423
Paterson ID, Mangan R, Downie DA, Coetzee JÁ, Hill MP, Burke AM, Downey PO, Henry TJ, Compton SG (2016) Two in one: cryptic species discovered in biological control agent populations using molecular data and crossbreeding experiments. Ecol Evol 6(17):6139–6150
Pellizzari G, Germain J-F (2010) Scales (Hemiptera, Superfamily Coccoidea). In: Roques A (ed) Alien terrestrial arthropods of Europe. BioRisk, pp 475–510
Pesson P (1941) Description du male de Pulvinaria mesembryanthemi Vallot et observations biologiques sur cette espèce (Hemipt. Coccidae). Ann Soc Entomol France 110:71–77
Pina-Martins F, Paulo OS (2008) Concatenator: sequence data matrices handling made easy. Mol Ecol Resour 8:1254–1255. https://doi.org/10.1111/j.1755-0998.2008.02164.x
Porco D, Potapov M, Bedos A, Busmachiu G, Weiner WM, Hamra-Kroua S, Deharveng L (2012) Cryptic diversity in the ubiquist species Parisotoma notabilis (Collembola, Isotomidae): a long-used chimeric species? PLoS ONE 7:e46056. https://doi.org/10.1371/journal.pone.0046056
Rambaut A, Drummond AJ, Xie D, Baele G, Suchard MA (2018) Tracer v1.6. http://tree.bio.ed.ac.uk/software/tracer/
Rauth SJ, Hinz HL, Gerber E, Hufbauer RA (2011) The benefits of pre-release population genetics: a case study using Ceutorhynchus scrobicollis, a candidate agent of garlic mustard. Alliaria petiolata Biol Control 56:67–75. https://doi.org/10.1016/j.biocontrol.2010.09.015
Rosen D (1986) The role of taxonomy in effective biological control programs agriculture. Ecosyst Environ 15:121–129. https://doi.org/10.1016/0167-8809(86)90085-X
Salisbury A, Malumphy C, Halstead AJ (2011) First incursions of Aloea australis (Hemiptera: Miridae) and Pulvinaria delottoi (Hemiptera: Coccidae) in Europe, and three other hemipteran insects imported from South Africa. Br J Ent Nat Hist 24:217–221
Santoro R, Jucker T, Carboni M, Acosta ATR (2012) Patterns of plant community assembly in invaded and non-invaded communities along a natural environmental gradient. J Veg Sci 23:483–494. https://doi.org/10.1111/j.1654-1103.2011.01372.x
Schroer S, Pemberton RW, Cook LG, Kondo T, Gullan PJ (2008) The genetic diversity, relationships, and potential for biological control of the lobate lac scale, Paratachardina pseudolobata Kondo & Gullan (Hemiptera: Coccoidea: Kerriidae). Biol Control 46:256–266. https://doi.org/10.1016/j.biocontrol.2008.04.009
Seljak G (2010) A checklist of scale insects of Slovenia. Entomol Hell 19:99–113
Sequencher® version 4.0.5 (2003) DNA sequence analysis software. Genes Code Corportation, Ann Arbor
Sethusa MT, Millar IM, Yessoufou K, Jacobs A, van der Bank M, van der Bank H (2014) DNA barcode efficacy for the identification of economically important scale insects (Hemiptera: Coccoidea) in South Africa. Afr Entomol 22(2):257–266
L. Swofford D (2002) PAUP*. Phylogenetic analysis using parsimony (*and other methods). Version 4.0b10 vol Version 4.0. https://doi.org/10.1111/j.0014-3820.2002.tb00191.x
Thompson JD, Higgins DG, Gibson TJ (1994) CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 22:4673–4680
Van Grunsven RHA, Bos F, Ripley BS, Suehs CM, Veenendaal EM (2009) Release from soil pathogens plays an important role in the success of invasive Carpobrotus in the Mediterranean. S Afr J Bot 75:172–175. https://doi.org/10.1016/j.sajb.2008.09.003
Vieira RMdS, Carmona MM, Pita MdS (1983) Sobre os coccídeos do Arquipélago da Madeira (Homoptera - Coccoidea). Boletim do Museo Municipal do Funchal 35:81–162
Wang XB, Deng J, Zhang JT, Zhou QS, Zhang YZ, Wu SA (2015) DNA barcoding of common soft scales (Hemiptera: Coccoidea: Coccidae) in China. Bull Entomol Res 105:545–554. https://doi.org/10.1017/s0007485315000413
Washburn JO, Frankie GW (1981) Dispersal of a Scale Insect, Pulvinariella mesembryanthemi (Homoptera: Coccoidea) on Iceplant in California. Environ Entomol 10:724–727. https://doi.org/10.1093/ee/10.5.724
Washburn J, Frankie G (1985) Biological studies of iceplant scales, Pulvinariella mesembryanthemi and Pulvinaria delottoi (Homoptera: Coccidae), in California. Hilgardia 53:1–27. https://doi.org/10.3733/hilg.v53n02p027
Acknowledgements
We thank Íñigo Sánchez García, Jesús R. Aboal Viñas, José Rafael González López, Kate McCombs, Serafín J. González Prieto and Stefan Neser for providing biological samples for this study. We are also grateful to Pilar Soengas for her suggestions to improve the paper and to Serafín J. González Prieto and the CoBIG2 group for their advice and assistance. The participation of Cristina Vieites-Blanco was supported by a pre-doctoral fellowship by Xunta de Galicia and an internship grant by Santander Universidades.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Vieites-Blanco, C., Paulo, O.S., Marabuto, E. et al. Genetic variability on worldwide populations of the scale insect Pulvinariella mesembryanthemi. Biol Invasions 22, 735–748 (2020). https://doi.org/10.1007/s10530-019-02125-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10530-019-02125-3