
Abstract
Understanding the population genetic diversity and structure of recently introduced pest species is important for determining appropriate strategies for pest control. In recent years, the rate of introduction of new invasive insect pests between continents has continued to increase. About a decade ago, the soybean aphid, Aphis glycines Matsumura, was introduced from East Asia (EA) into North America (NA) and is now widely established in NA. To compare soybean aphid populations between the native and invasive regions, we examined 689 individuals obtained from 28 different collections in NA and EA. A total of 8 microsatellite loci were used for population genetics statistics. Genetic differentiations among NA populations were very low compared to those among EA populations. Gene diversity and mean number of alleles in NA populations averaged 0.40 and 2.70, respectively, whereas in EA they averaged 0.55 and 4.32, respectively. Structure analysis of all populations revealed two distinct structures in the invaded and in the native regions. NA populations were divided into two distinct structures consistent with their geographic distribution. Among EA populations, certain Korean populations were genetically closest to NA populations, especially those from Ohio and Delaware. An approximate Bayesian computation test also supports an introduction into NA from Korea. These suggest that Korea is most likely to be the origin of soybean aphids in North America, rather than China or Japan.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Invasions of exotic species often result in serious damage to indigenous biodiversity, ecosystems, public health, and agricultural productivity in the introduced regions when the species successfully establish (Sax et al. 2005). As international trade expands, the probability of introduction of exotic insect species between continents increases (Dalmon et al. 2008; Lozier et al. 2009). For phytophagous insects, the food source or host as well as climatic conditions are major factors for successful survival and spread in the introduced regions. Insect pests that damage agricultural or economic plants can easily adapt to the introduced regions which already have suitable environments such as agricultural crop fields. The outbreaks that often follow biological invasions frequently cause serious damage in agriculture, which is only part of their total economic impact (Miller et al. 2005).
Due to climatic similarities between North America and East Asia, transpacific introductions of exotic pests have been increasing in both regions. Recently, the brown marmorated stink bug, Halyomorpha halys (Stål), was introduced from Asia to North America where it is expected to cause serious damage to the soybean economy along with the soybean aphid (Zhu et al. 2012). Wood boring larvae of the Asian longhorn beetle, Anoplophora glabripennis (Motschulsky), which seriously damage trees and wood materials in North America are native to China and Korea (Carter et al. 2010). Conversely, the citrus flatid planthopper, Metcalfa pruinosa Say, recently invaded from North America and has dispersed throughout Korea and damages many fruit and ornamental trees (Kim et al. 2011). Because of the environmental similarity of these two regions and the continuation of trade activities, it is expected that the invasion of exotic insects across the Pacific will increase in the future.
Interpreting the population genetic diversity and structure of recently introduced pest species is important for establishing strategies for pest control. Population genetic studies can be useful to monitor the migration of insects and to confirm resistant or susceptible strains for sustaining insect-resistant crop cultivars (Llewellyn et al. 2003; Yencho et al. 2000). Another important aspect of dealing with invasive species is the determination of their origin (Guillemaud et al. 2010). This knowledge supports a strategy of biological control by the introduction of natural enemies (Desneux et al. 2009; Torchin et al. 2003). By comparing the genetic structure of source and invasive populations, the origin can often be identified by using various genetic markers. In particular, microsatellite markers have been used in many studies aimed at clarifying the origins of invasive species (Guillemaud et al. 2010; Miller et al. 2005). Microsatellites are not only reliable for studying recent biological invasions (Zygouridis et al. 2009), but are also powerful enough to detect the origin and invasion route of exotic insect species (Fonseca et al. 2010). Moreover, using multilocus genotypes based on a combination of multiple loci, researchers are able to precisely identify the migration route between source and invasive populations (Paetkau et al. 2004; Wilson and Rannala 2003). As a representative case, Miller et al. (2005) revealed that the western corn rootworm, Diabrotica virgifera virgifera LeConte, was introduced into several European regions from North America through at least three independent transatlantic invasions.
Microsatellites have been widely used in many aphid studies based on population genetics, including genotypic diversity (Kanbe and Akimoto 2009), biological invasion (Lozier et al. 2009), lineage of asexuality (Delmotte et al. 2002, 2003), host range expansion (Peccoud et al. 2008), ecological specialization (Carletto et al. 2009), and speciation (Lozier et al. 2007). Due to an unusual biological characteristic of aphids, i.e. cyclically or entirely parthenogenesis, microsatellites are ideally suited for examining heritable genetic variation and genetic evolution on aphids (Wilson et al. 2003). In addition, because a metapopulation of a polyphagous species specifically adapted to one host plant tends to keep reproducing continuously by parthenogenesis at moderate temperatures, population structures can be clearly identified using microsatellites between colonies on different host plants, even for sympatric populations (Carletto et al. 2009; Peccoud et al. 2009). Based on this suitability for application on aphids, microsatellites have now been intensively developed in major aphid groups such as Aphidinae, Calaphidinae, and Eriosomatinae (Lavandero et al. 2009; Wilson et al. 2004; Yao et al. 2003). For aphidologists, population genetics analysis of aphids can provide much useful evidence with regard to migration, dispersal, geographic variation, and evolution of host associations.
Population genetics analysis using microsatellite loci has made it possible to trace the origin, route of invasion, and dispersal pattern of introduced aphids. Population genetic studies of invasive aphids successfully revealed the origin and invasion route of several introduced aphids considered among the world’s most notorious pests. Lozier et al. (2009) provided evidence for multiple invasions of the mealy plum aphid, Hyalopterus pruni (Geoffroy), into North America from several nonadjacent European regions. Margaritopoulos et al. (2009) confirmed genetic relationships and variations of the peach-potato aphid, Myzus persicae (Sulzer), which is widespread throughout the world, based on global scale sampling. Zepeda-Paulo et al. (2010) studied the invasion of Myzus persicae nicotianae (Blackman), which recently diverged from M. persicae, in tobacco fields of South America, (Blackman and Spence 1992). It was revealed that this aphid recently colonized South America subsequent to its invasion of North America and not directly from its native range in Europe or Asia (Zepeda-Paulo et al. 2010).
In general, aphids have shown the ability to successfully adapt and disperse when they are introduced into new regions because of their host specific associations and parthenogenetic reproduction. They are easily transferred due to the uniform environment of agricultural fields which is extensively developed and structured. Population genetic studies of invasive aphids often show low genetic variation in the introduced region shortly after the introduction (Harrison and Mondor 2011; Zepeda-Paulo et al. 2010). Multilocus genotypes of invasive aphids based on multiple microsatellite loci generally exhibited a lack of genotypic diversity. The low genetic variation was derived from their cyclic or permanently parthenogenetic life rather than from a primary genetic bottleneck. In addition, having only one sexual phase per year reduces the chance of genetic heterogeneity, and some pest species such as the cotton-melon aphid, Aphis gossypii Glover, and the peach-potato aphid, M. persicae, usually omit the sexual phase under greenhouse conditions (Blackman and Eastop 2006), which also was even discovered in cold regions. Therefore, genetic characterization using microsatellites enables population genetics analysis to more clearly confirm the origin and route of invasion of aphids than for other insects with obligatory sexuality.
The soybean aphid, Aphis glycines Matsumura, was recently introduced into North America where it has become a serious pest of soybeans (Venette and Ragsdale 2004). This invasive pest rapidly spread throughout the north central United States and southern Canada since its discovery in 2000 (Ragsdale et al. 2011). The soybean aphid has an obligatory heteroecious and holocyclic life cycle (Wu et al. 2004). The aphids live on the secondary (summer) host, the soybean, during late spring, summer and early fall seasons, then migrate to the primary (winter) host, buckthorn (Rhamnus). Four buckthorn species, Rhamnus alnifolia L’Hér., R. cathartica L., R. davurica Pall., and R. japonica Maxim. have been confirmed as suitable primary hosts in East Asia and North America (Kim et al. 2010). Among them, the European species R. cathartica is considered to be the major primary host of the soybean aphid in North America (Heimpel et al. 2010). Because of such primary and secondary host relationships, the question has often been posed whether or not the primary host contributes to genetic transmission between different aphid populations. However, the population genetic structures and differentiation between primary (Rhamnus) and secondary (soybean) hosts are still unknown.
At this time, the origin of invasive populations of A. glycines in North America has not yet been confirmed. Venette and Ragsdale (2004) suggested that Japan could have been the source of the infestation due to a favorable climate match with North America. The first population genetics studies of soybean aphid were recently performed using cross-amplifiable microsatellite loci developed from other Aphis species (Michel et al. 2009a, b). After comparing 2 South Korean and 10 North American populations, Michel et al. (2009b) suggested that South Korea could be excluded as the source of the North American invasion. However, the allelic polymorphisms reported were too low to explain the soybean aphid’s genetic diversity (Michel et al. 2009b). To interpret the invasion history of soybean aphid, additional polymorphic markers isolated from soybean aphid were needed. Meanwhile, 10 novel microsatellite markers were isolated and developed from soybean aphid, which was quite polymorphic from within a single colony of individuals (Andree et al. 2010). It was expected that these loci would facilitate research on the population genetics of the soybean aphid.
Our objective was to compare the population genetic diversities and structures of the invasive region, North America (USA), and the native region, East Asia (Korea, China, and Japan), focusing mainly on Korea, using these recently developed microsatellites (Andree et al. 2010). We characterized the genetic diversity of soybean aphid in these regions, and compared the genetic differentiations between the regional populations. We also inferred the most likely source population for the North American invasive populations by using Bayesian inference methods.
Methods
Soybean aphid samples and identification
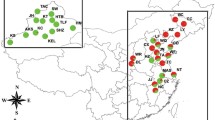
We examined 689 individuals obtained from 28 different collections, including the regional collection sites in North America and East Asia (Fig. 1; Table 1). Of these, 8 collections were acquired from USA and 20 collections were from Korea, China, and Japan. The latitude of East Asian collections approximates that of North America collections. To examine the genetic diversity between primary and secondary host populations and between field and laboratory populations, 5 collections were obtained from primary hosts, buckthorn species (R. cathartica in North America and R. davurica in East Asia), 3 collections from laboratory cultures, and the remaining 20 from soybean fields. Laboratory populations were collected from three different locations in All rearing were done in an environmental room that was maintained at 25 ± 0.5 °C, 50 ± 0.3 % relative humidity, and 16 h light: 8 h dark (LD 16:8) photoperiod. To avoid the chance of sampling individuals from the same parthenogenetic colony, each aphid was collected from a different host plant (soybean) or from a different isolated colony (on buckthorn). All of the fresh aphid specimens used for molecular analyses were collected and preserved in 95 or 99 % ethanol and stored at −70 °C. On the primary host, Rhamnus sp., soybean aphids could coexist with several morphologically similar Aphis species that have been relatively little-studied for diagnostic characteristics. Therefore, we applied the molecular identification method (Kim et al. 2010) using two mitochondrial regions, the COI barcode region and the partial tRNA-leucine + COII, for which 8 North American and 20 East Asian representative individuals were identical in a total of 1360 bp sequences (data not shown).

Approximate locations of collection sites: a USA, b Korea, c East China and Japan
Microsatellite genotyping
In this study, all 689 individuals were genotyped using eight microsatellite loci (AGL1-2, AGL1-10, AGL1-11, AGL1-15, AGL1-16, AGL1-21, AGL1-22, and AGL2-3b) previously isolated from the soybean aphid (Andree et al. 2010). Although the two remaining (AGL1-20 and AGL2-6) loci developed by Andree et al. (2010) were also used, these loci were not included in our analyses because they were uninformative due to prevailing monomorphisms in most population samples.
Total genomic DNA was extracted from single individuals using a DNeasy® Blood & Tissue Kit (QIAGEN, Inc., Dusseldorf). For preserving voucher specimens from the DNA extracted samples, we used a non-destructive DNA extraction protocol (Kim et al. 2010). The entire body of the aphid was left in the lysis buffer with protease K solution at 55 °C for 24 h and the cleared cuticle dehydrated. Microsatellite amplifications were performed using GeneAll® Taq DNA Polymerase Premix (GeneAll, Seoul, Korea) in 20 μl reaction mixtures containing 0.5 μM forward labeled with a fluorescent dye (6-FAM, HEX, or NED) and reverse primers and 0.05 μg of DNA template. PCR was performed using a GS482 thermo-cycler (Gene Technologies, Essex) according to the following procedure: initial denaturation at 95 °C for 5 min, followed by 34 cycles of 95 °C for 30 s; annealing at 56 °C for 40 s; extension at 72 °C for 45 s, and a final extension at 72 °C for 5 min. PCR products were visualized by electrophoresis on a 1.5 % agarose gel with low range DNA ladder to check for positive amplifications. Automated fluorescent fragment analyses were performed on the ABI PRISM 377 Genetic Analyser (Applied Biosystems), and allele sizes of PCR products were calibrated using the molecular size marker, ROX labeled-size standard (GenScan™ ROX 500, Applied Biosystems). Raw data on each fluorescent DNA product were analyzed using GeneMapper® version 3.7 (Applied Biosystems).
Data analysis
We used GENCLONE 2.0 (Arnaud-Haond and Belkhir 2007) to identify multilocus genotypes (MLGs) among populations and to calculate the genotypic diversity (GD = [G/N]), where G is the number of different MLGs and N is the sample size (Dorken and Eckert 2001). Observed (H O) and expected heterozygosity (H E) values among loci were estimated using GENEPOP 4.0.7 (Raymond and Rousset 1995) among the population data sets as well as between North American and East Asian data sets. Levels of significance for Hardy–Weinberg equilibrium (HWE) and linkage disequilibrium tests were adjusted using the sequential Bonferroni correction for all tests involving multiple comparisons (Rice 1989). Deviations from HWE were tested for heterozygote deficiency or excess. MICRO-CHECKER (Oosterhout et al. 2004) was used to test for null alleles (Brookfield 1996) and identify possible scoring errors because of the large-allele dropout and stuttering. The program FSTAT 2.93 was used to estimate the gene diversity (H S), mean number of alleles (N A), and allelic richness (R S).
We also used FSTAT for calculations of pairwise genetic differentiation (F ST) values (Weir and Cockerham 1984), in which populations were assigned by location. Two or three populations collected from same location were grouped as one geographical population for DE, SD, HS, SW, and OS, except for one laboratory population (SW-L). We subsequently used 18 geographical populations in other analyses. The statistical significance of each value was assessed by the comparison of the observed value with the values obtained in 153,000 matrix permutations.
To examine genetic relationships between individual samples of A. glycines, the factorial correspondence analysis (FCA) in GENETIX version 4.05 (Belkhir et al. 2005) further explored population relationships using the microsatellite data, making no a priori assumptions about population groupings.
The program STRUCTURE 2.3.3 (Pritchard et al. 2000) was used to test for the existence of population structuring among all samples, among North American, and among East Asia by estimating the number of distinct populations (K) present in the set of samples using a Bayesian clustering approach. We assessed likelihoods for models with the number of clusters ranging from K = 1 to K = 15. The length of the initial burn-in period was set to 100,000 iterations followed by a run of 1,000,000 Markov chain Monte Carlo (MCMC) repetitions, replicated 10 times to ensure convergence on parameters and likelihood values. Parameter sets of ancestry, allele frequency, and advanced models were remained as defaults. Following the method of Evanno et al. (2005), we calculated ΔK based on the second order rate of change in the log probability of data with respect to the number of population clusters from the STRUCTURE analysis. From the value of ΔK = 2, population substructure was indicated. We chose the run with the greatest likelihood at K = 2 to assign individuals into two subpopulations and we analyzed those groups separately in STRUCTURE to determine the correct value of K.
We performed assignment tests using GENECLASS 2 (Piry et al. 2004) in which populations were assigned to 18 geographical populations. The program calculates, for each individual of a population, the probability of belonging to any other reference population or to be a resident of the population where it was sampled. The sample with the highest probability of assignment was considered the most likely source for the assigned genotype. We used a Bayesian method of estimating population allele frequencies (Rannala and Mountain 1997). Monte Carlo re-sampling computation (10,000 simulated individuals) was used to infer the significance of assignments (alpha = 0.01).
Groupings based on biogeography, host associations, and field versus lab collections were tested independently with analysis of molecular variance (AMOVA; Excoffier et al. 2005) in ARLEQUIN, with significance determined using the nonparametric permutation approach described by Excoffier et al. (1992).
The program BOTTLENECK version 1.2.02 (Piry et al. 1999) was used to detect the effect of a recent bottleneck in all populations in our samples. Two mutation models, considered appropriate for microsatellites (Piry et al. 1999), were applied as the strict Stepwise Mutation Model (SMM) and the Two-phase Model (TPM). For the TPM, a model that includes both 90 % SMM and 10 % TPM was used for 20,000 iterations. Significant deviations in observed heterozygosity over all loci were tested using a nonparametric Wilcoxon sign-rank test (Cornuet and Luikart 1996).
To estimate the relative likelihood of alternative introduction scenarios of the soybean aphid, an approximate Bayesian computation (ABC) was performed for microsatellite data as implemented in DIYABC version 1.0.4 (Cornuet et al. 2008). DIYABC allows the comparison of complex scenarios involving bottlenecks, serial or independent introductions, and genetic admixture events in introduced populations (Estoup and Guillemaud 2010). The parameters for modelling scenarios are the times of split or admixture events, the stable effective population size, the effective number of founders in introduced populations, the duration of the bottleneck during colonization, and the rate of admixture (Cornuet et al. 2010). The software generates a simulated data set used to estimate the posterior distribution of parameters in order to select the most likely scenario (Cornuet et al. 2010). DIYABC generates a simulated data set that is then used to select those most similar to the observed data set, and so called selected data set (n δ ), which are finally used to estimate the posterior distribution of parameters (Cornuet et al. 2008).
It was necessary to test two different ABC analyses using all or partial datasets. Because we did not know which location is the most ancestral population of the soybean aphid among Korea (except for laboratory populations), China, and Japan, we first performed the ABC analysis to confirm that using only Asian population samples except for USA ones. Three different scenarios (A1, A2, and A3) were set as follows. Scenario A1 considered Korea and Japan originated from China. Scenario A2 was China and Japan originated from Korea. Scenario A3 was China and Korea originated from Japan. We then performed the second ABC analysis to determine the most likely source of introduction among three Asian countries, hypothesizing three scenarios. As a result of the first analysis, China was the most ancestral population among them (see Results), thereafter China was set to the most ancestral (basal) population. In the second test, three different scenarios (B1, B2, and B3) were set as follows. Scenario B1 was a clade of Korea and Japan diverged from China and then USA introduced from Korea (source). Scenarios B2 was a clade of Korea and Japan diverged from China and then USA introduced from Japan (source). Scenarios B3 was a clade of Korea and Japan diverged from China and then USA introduced from China (source).
We produced 1,000,000 simulated data sets for each scenario. We used a generalized stepwise model (GSM) as the mutational model for microsatellites, which assumes increases or reductions by single repeat units (Cornuet et al. 2008). To identify the posterior probability of these three scenarios, the n δ = 30,000 (1 %) simulated datasets closest to the pseudo-observed dataset are selected for the logistic regression, which are similar to the n δ = 300 (0.01 %) ones for the direct approach (Cornuet et al. 2010). The summary of statistics was calculated from the simulated and observed data for each of the tested scenarios such as mean number of alleles per locus (A), mean genetic diversity for each group and between group, genetic differentiation between pairwise groups (F ST ), classification index, shared alleles distance (D AS ) and Goldstein distance.
Results
Genetic diversity within populations
We successfully genotyped 689 soybean aphid individuals using 8 microsatellite loci and found 505 non-clonal MLGs from all samples (Table 2). Overall genotypic diversity (GD) averaged 0.74, but very low GD (0.30–0.50) was detected in three laboratory (SW-L, CW-L, NS-L) and one field (OM) populations. There was no evidence of significant linkage disequilibrium or frequency of null alleles. Expected heterozygosity (H E) values in East Asian populations ranged from 0.353 to 0.680, whereas H E values ranged from 0.292 to 0.478 in North American populations. In HWE, there were significant deviations in OH, SD3, SW-L, CW-L, NS-L, and OM by heterozygote excess, and in HS1, OS2, YT, QD, and SH by heterozygote deficit. Heterozygote excess in three field populations (OH, SD3, JP) likely were the result of heterosis or over-dominance related to selection preference toward heterozygous combination (Delmotte et al. 2003). In contrast, heterozygote excess in three laboratory populations (SW-L, CW-L, NS-L) seemed to be caused by the fixation of heterozygous genotypes because H O values also were largely skewed in laboratory populations (Michel et al. 2010). Homozygote excess in two Korean populations (HS1, OS2) likely was due to retaining numerous unique genotypes with private alleles within a population related to their relatively high H E. Genetic diversity was generally high throughout the native region. Gene diversity (H S) and mean number of alleles (N A) in North American populations averaged 0.40 and 2.70, respectively, whereas East Asian populations averaged 0.54 and 4.38, respectively. Similarly, allelic richness (R S, mean ± S.D., 2.12 ± 0.11) in North American populations were much lower than R S (2.99 ± 0.68) in East Asian populations.
Genetic differentiation between populations
We estimated pairwise genetic differentiation (F ST) between 18 different geographical populations (Table 3). For the analyses, several collections obtained from same or adjacent location were merged into one geographical population (e.g. DE1 and DE2 into DE). Pairwise comparisons of F ST values showed that North American populations were genetically closer to Korean field populations than Chinese or Japanese populations. F ST between North American and Korean field populations averaged 0.258, whereas F ST between North American and Chinese and Japanese populations averaged 0.387 0.369, respectively. Three laboratory populations in Korea were genetically more distant from North American populations, with F ST ranging from 0.342 to 0.564, than East Asian field populations. F ST estimates within North American populations, which averaged 0.096, were much lower than those within Korean (or Chinese) field populations, which averaged 0.258 (0.250). This corresponds to relatively low gene and allelic diversities of North American populations as shown in Table 1. Surprisingly, F ST values between OH and five Korean populations (HS, SW, OS, GJ, MY) were relatively low (averaging 0.139), which was lower than F ST between DE and IL (0.173).
Genetic structure and assignment
A plot of FCA showed that North American and East Asian samples were completely separated in each of the right and left sides on the plot (Fig. 2). Individual plots of North American samples were closely aggregated along the line of factor 1, whereas the plots of East Asian samples were relatively scattered. Among all East Asian populations, only a few Korean samples were located near North America samples. Chinese and Japanese samples were located further from North American samples than Korean samples.

Plot of the FCA based on the first two factors for 689 individuals of the soybean aphid
In all STRUCTURE analyses, the most likely number of clusters was K = 2 using the ΔK calculation (from K = 1 to K = 15). The STRUCTURE analysis for all samples resulted in two distinct clusters, North American and East Asian populations (Fig. 3a). However, the green cluster appeared to a slight degree in DE1, DE2, and OH, and the red cluster was sparsely distributed among Korean populations, especially in OS2, GJ and MY. Chinese (YJ) and Japanese (OM) populations barely had the red cluster compared with the Korean populations. The individual STRUCTURE analyses for each of North American and East Asian populations showed that there were two conspicuous genetic clusters in each region (Fig. 3b). In North America, one cluster (in blue) was prevalent in DE and OH, and, a second (in yellow) was dominant in IN, IL, and SD. In East Asia, the dark blue cluster was predominantly distributed in HC, YJ, YT, and YC, and the pale yellow cluster appeared in YE, QD, and OM, whereas most of the Korean locations showed mixed clusters.

Genetic structure of the soybean aphid a for all samples (K = 2) and b for each of North American and East Asian populations individually (both K = 2). The increase of the likelihood value when K = 2 was highest using ΔK calculation for all tests. Lettered location labels correspond to Table 1
The Bayesian assignment tests using GENECLASS 2 were carried out to identify population membership of individuals from all populations. The result of the assignment test (Table 4) indicated the average probability with which individuals were assigned to the corresponding reference population. The self-assignment probability values averaged 0.468 ± 0.05 (mean ± S.D.) in North American populations, and 0.540 ± 0.14 in East Asian populations except laboratory populations. Laboratory populations in Korea had high self-assignment rates (0.850 ± 0.05). The populations collected from the primary host (Rhamnus spp.) had relatively low self-assignment rates (0.376 ± 0.05). The assignment approach also could be used to interpret the inference of invasion of soybean aphid genotypes into North America from East Asia. All North American populations (DE, OH, IN, IL, SD) could be assigned to five Korean populations (HS, SW, OS, GJ, MY), but no Chinese and Japanese populations were assigned to the North American populations. In particular, OH showed a high mean assignment rate for OS (0.489), which is approximately its self-assignment rate (0.533). This strongly suggests that North American populations originated from Korea rather than other East Asian regions.
Genetic variance and bottleneck
Eight cases to confirm genetic variance between the preordained groups were analyzed using AMOVA implemented in ARLEQUIN (Excoffier et al. 2005). Due to relative lack of Chinese and Japanese populations, genetic variance among groups in both case 1 (between USA and East Asia) and case 2 (between USA and Korea) were similar at about 18 %. However, genetic variance of about 25–30 % among groups in both case 3 (between USA and China) and case 4 (between USA and Japan) suggests that there are relatively different regional structures between the compared regions. Other cases (5, 6, 7, 8) showed that the majority of genetic variation was generally found among populations within groups and among individuals within populations. No significant genetic variation existed between primary and secondary host-associated populations. Genetic variance among groups due to laboratory isolation in Korea was somewhat higher than genetic variance between groups DE + OH and IN + IL + SD, which showed different genetic structural composition in the structure analysis.
Although the bottleneck test should be cautiously regarded because sample size for some populations was less than 30 individuals (Cornuet and Luikart 1996), there was no evidence of a recent population bottleneck in any of the populations. In our study, genetic diversity of North American populations was shown to be much lower than that of native populations. It is doubted that NA populations have undergone a genetic bottleneck after invasion from EA and bottleneck software failed to find evidence of a genetic bottleneck.
Inferring an introduction to test hypothetical scenarios by ABC analysis
To propose the invasive scenario with the greatest likelihood, followed by the soybean aphid when introduced into USA, two ABC tests were conducted (Figure S1). The first one included the dataset from only Asian populations testing for three scenarios to determine which location is the most ancestral among three countries (China, Korea, and Japan). These results are presented as a logistic regression using DIYABC software, estimating the posterior probability of each tested evolutionary scenarios of invasion for the selected simulated data (n δ ) (Cornuet et al. 2008), which ranged between 3000 and 30,000 n δ . In the first analysis, ABC simulations based on three alternative scenarios (A1, A2, and A3) for origin of the soybean aphid gave strong support to scenario A1 (mean posterior probability: 0.97–0.99; 95 % CI = 0.99–0.99 to 0.97–0.98) and similar posterior parameter estimates, independent of the sets (A2 and A3) used. The two other scenarios showed much lower posterior probabilities (<0.03). As the result, China was set to the most original (basal) population in the following analysis. The second analysis tested for three scenarios (B1, B2, and B3) hypothesizing an introduction from one of the Asian countries. In this analysis, scenario B1 obtained the highest posterior probability ranging from 0.90 (n δ = 3000) to 0.91 (n δ = 30,000) with a 95 % CI of 0.81–0.99 and 0.88–0.94, which assumes an introduction from Korea. Scenario B2, which assumes an introduction from Japan, showed a posterior probability ranging from 0.07 (n δ = 3000) to 0.06 (n δ = 30,000) with a 95 % CI of 0–0.15 and 0.04–0.08. Scenario B3, which assumes an introduction from China, showed a posterior probability ranging from 0.03 (n δ = 3000) to 0.03 (n δ = 30,000) with a 95 % CI of 0–0.07 and 0.02–0.04. These suggest that scenario B1 appeared as the most robust hypothesis, presenting the highest posterior probability of the three scenarios tested.
Discussion
Genetic diversity and structure of soybean aphid populations between North America and East Asia
The invasive populations of soybean aphid in North America appear to have lower genetic diversity than the source populations in East Asia. Comparing mean number of alleles (N A) or allelic richness (R S) between North American and East Asian populations, the former’s indices were relatively low, limited mainly by a low number of alleles. In addition, genotypic diversity (GD) restricted by lack of alleles in North America also was generally lower than in East Asia. Genetic differentiations (F ST) among North American populations were very low compared to F ST among Korean populations even although some Korean populations are closely placed to North American populations. (Michel et al. 2009b) suggested that the soybean aphid invasion in North America was initiated from a pattern of a small colonizing populations, followed by rapid clonal amplification and subsequent large-scale differentiation (Ragsdale et al. 2011). Genetic variations and differences between North American populations can be simply explained by the FCA result (Fig. 2) which shows that North American soybean aphids are still genetically close. Thus, our results suggest that the initial founders of soybean aphid populations in North America were not genetically diverse, i.e. soybean aphids probably came from a single source population rather than from many locations by multiple introductions.
There were two apparently different genetic structures between North America and East Asia based on STRUCTURE and AMOVA analyses (Fig. 3; Table 5). Our study showed that the populations in the invasive and source regions have genetically diverged. In the bottleneck analysis, significant genetic bottlenecks were not detected. However, two genetically independent structures in STRUCTURE had a few mixed patterns, and FCA (Fig. 2) showed the existence of a genetic gap between North American and East Asian individuals probably caused by a genetic bottleneck effect, even though it was not detected. This suggests that North American populations of soybean aphid have undergone a founder effect soon after their invasion, and subsequently evolved novel allelic patterns different from the original ones, recovering some allelic diversity and frequency. The reason the bottleneck analysis was not positive may be explained by the North American populations of the soybean aphid having obtained a sufficient gene pool independently from their original range over several mating seasons.
Effect of host alternation on genetic diversity and differentiation
Unlike most aphids which are monoecious (Blackman and Eastop 1994, 2006), Aphis glycines is a heteroecious holocyclic aphid. One monoecious species, the pea aphid, Acyrthosiphon pisum Harris, generally has both asexual and sexual lineages, and some asexual populations showed drastic change and a decrease in their allele diversity and frequency in contrast to sexual lineages (Kanbe and Akimoto 2009). In addition, it was found that different host preferences could cause incipient speciation driven by host-specific isolation (Peccoud et al. 2009). In contrast, all soybean aphids are obligatorily heteroecious. Thus, soybean aphids have one opportunity per year to exchange alleles and vary genotypes through sexual exchange among various populations on the primary (winter) host. Furthermore, it was recently found that an increase in genetic diversity could occur from late-season dispersal within soybean before migration to the winter host, Rhamnus (Orantes et al. 2012).
Host alternation between primary and secondary hosts may contribute to increasing genetic diversity. Soybean aphid populations mix their alleles once a year when they during mating on the winter host (Heimpel et al. 2010; Kim et al. 2010). Correspondingly, our data showed that genotypic diversity (GD) in populations on buckthorn were somewhat higher than in populations on soybean. This was especially true for OS2, one of buckthorn populations in Korea, which had the highest values in both mean number of alleles (N A) and allelic richness (R S). Overall, however, there were no significant genetic differences between buckthorn and soybean populations (Table 5). It was recently confirmed that soybean aphids could form a genetically correlated spatial distribution on the scale of ~300 km (Orantes et al. 2012). This suggests that soybean aphids are able to migrate relatively long distances. Thus, assuming the process of locating the primary host is not difficult, gene flow may be not restricted and genetic structure not distorted by seasonal host alternation.
Some monoecious anholocyclic and few completely asexual aphids often exhibit reduced, simple genetic diversity in their invasive locations. One example is the pest Myzus persicae nicotinae, which was introduced from Europe into North and South America (Zepeda-Paulo et al. 2010). Another example is the monoecious species, Aphis nerii, which typically feeds on oleander and milkweed, and which was also introduced into North America from Europe (Harrison and Mondor 2011). Interestingly, after investigation of their genotypes, both aphid species showed clonal effect on genetic variations in their invasive range (Harrison and Mondor 2011; Zepeda-Paulo et al. 2010). Spread of a dominant genotype which has a high ability for ecological adaptation tends to render genotypic variation simple in aphids (Harrison and Mondor 2011; Zepeda-Paulo et al. 2010). Because those a few genotypes reproduce asexually, their genotypes can be fixed, allowing them to dominate widely. This biological feature makes their allelic diversity simpler than in host-alternating species.
Relationships between known biotypes and genetic structures in North America
The STRUCTURE analysis (Fig. 3b), divided North American populations into two different genetic structures, with each location tending to have one dominant structure. Surprisingly, the structures of both DE and OH indicated dominance of the “blue” attribute, but those of IN, IL, and SD showed dominance of the “yellow” attribute. The presence of two soybean aphid biotypes has been previously confirmed in North America (Hill et al. 2010; Kim et al. 2008). The aphid isolate collected in Ohio was tested on soybean aphid resistant soybean lines, including lines with Rag1, and could be distinguished from an Illinois isolate by its ability to colonize plants with Rag1 (Kim et al. 2008). With the possibility that additional soybean aphid biotypes may be found in Illinois and Ohio, the Illinois aphid isolate was referred to as biotype 1 and the Ohio isolate biotype 2 (Hill et al. 2009). Interestingly, these results are consistent with our results in which OH could be distinguished from IL by STRUCTURE. Therefore, the microsatellite loci used in this study may also be useful to identify soybean aphids that are related to resistance or susceptibility to future soybean cultivars.
Inferring the origin of North American populations
The biological history of soybean aphid in North America began about a decade ago. In North America, the first infestation of aphid colonies on soybean was detected in southeastern Wisconsin in July, 2000 (Ragsdale et al. 2004). Venette and Ragsdale (2004) inferred that the invasive populations of soybean aphid in North America may have originated from Sapporo, Hokkaido, Japan, because this area was the closest climatic match among East Asian regions with North America, and also due to the frequent interceptions on imported materials from Japan.
Michel et al. (2009b) suggested that South Korea was not the source of the North American invasion based on comparison of 10 North American populations and 2 Korean populations. In his study, F ST estimates between two Korean populations (Hongcheon, Suwon) and USA populations were relatively large, averaging 0.361 (Michel et al. 2009b). Surprisingly, this genetic differentiation is similar to F ST values (averaging 0.366) between OH and three Korean laboratory populations in our study. We suggest two possibilities for why the previous study resulted in a large genetic differentiation between North American and Korean populations. The first is that Michel et al. (2009b) might have used Korean samples collected from laboratory populations or artificially isolated populations. It was shown that laboratory populations had a large genetic difference from field populations his subsequent study (Michel et al. 2010). Our study confirmed that there are significant differences between field and laboratory populations (Tables 3, 5). The F ST between Hongcheon and Suwon populations of 0.475 in the previous study of Michel et al. (2009a) is very similar to the F ST (0.483–0.498) between our laboratory populations. The second possibility is a lack of genetic resolution due to having used seven microsatellite loci developed from related species, Aphis fabae and A. gossypii (Coeur d’Acier et al. 2004; Gauffre and Coeur d’acier 2006; Vanlerberghe-Masutti et al. 1999) rather than from A. glycines. These markers were not polymorphic and had only two alleles (Michel et al. 2009a, b). In this condition of low allelic diversity, estimation of genetic differentiation on the basis of loss or gain of some alleles must be very sensitive. Thus, the estimates using cross-amplification markers may have led to low genetic diversity within the USA populations and large genetic differences between the USA and Korean populations.
In this study, Chinese and Japanese populations do not seem to be source ones of the introduction into USA based on the results. Although we were not able to include many Chinese and Japanese samples, regional populations unsampled in each country may be predicted to be close to our representative populations. The all Chinese populations (YJ, YT, QD, YC, SH) also differed to a large degree. The Japanese population (OM) was collected from Omachi city, Nagano, Honshu, Japan, which is geographically isolated by sea and about 800 km far from south of Sapporo, Kyushu which is predicted to the origin of introduction so far. This Japanese population also had a larger genetic difference from North American populations than all the Korean populations. Consequently, our data suggests that soybean aphid populations of Korea, especially those close to OS and MY, are most likely to be the origin of North American populations rather than Chinese or Japanese populations. Based on the population genetics statistics, OS and MY in Korea were genetically closest to DE and OH among North American populations. Considering that the F ST between OH and IL populations was 0.160, the F ST averaging 0.127 between OH and OS/MY is relatively small. In addition, the result of the GENECLASS 2 assignment test also indicated that OH could be assigned to OS at a relatively high mean rate (0.489) among North American populations.
Furthermore, ABC analysis revealed that one hypothetical scenario (B1), which assumes an introduction of the soybean aphid into USA from Korea, was very robustly suggested than the other two scenarios (B2, B3; see Results). This ABC analysis can corroborate traditional methods (e.g. GENECLASS 2) suggesting a possible history of the introduction on the basis of estimating evolutionary changes between native and invasive populations. As seen in all the results of the data analyses, Korea is the most likely source of invasive populations of the soybean aphid introduced into USA.
References
Andree K, Axtner J, Bagley MJ et al (2010) Permanent genetic resources added to molecular ecology resources database 1 April 2010–31 May 2010. Mol Ecol Res 10:1098–1105
Arnaud-Haond S, Belkhir K (2007) GENCLONE: a computer program to analyse genotypic data, test for clonality and describe spatial clonal organization. Mol Ecol Notes 7:15–17
Belkhir K, Borsa P, Chikhi L et al (2005) GENETIX 4.05, logiciel sous windows TM pour la génétique des populations. Laboratoire Génome, Populations, Interactions, CNRS UMR 5000, Université de Montpellier II, Montpellier (France)
Blackman RL, Eastop VF (1994) Aphids on the world’s trees: an identification and information guide. CAB International, Wallingford
Blackman RL, Eastop VF (2006) Aphids on the world’s herbaceous plants and shrubs, the aphids, vol 2. Wiley, Chichester
Blackman RL, Spence JM (1992) Electrophoretic distinction between the peach-potato aphid, Myzus persicae, and the tobacco aphid, M. nicotianae (Homoptera, Aphididae). B Entomol Res 82:161–165
Brookfield JFY (1996) A simple new method for estimating null allele frequency from heterozygote deficiency. Mol Ecol 5:453–455
Carletto J, Lombaert E, Chavigny P et al (2009) Ecological specialization of the aphid Aphis gossypii Glover on cultivated host plants. Mol Ecol 18:2198–2212
Carter M, Smith M, Harrison R (2010) Genetic analyses of the Asian long horned beetle (Coleoptera, Cerambycidae, Anoplophora glabripennis), in North America, Europe and Asia. Biol Invasions 12:1165–1182
Coeur d’Acier A, Sembene M, Audiot P et al (2004) Polymorphic microsatellites loci in the black Aphid, Aphis fabae Scopoli, 1763 (Hemiptera, Aphididae). Mol Ecol Notes 4:306–308
Cornuet JM, Luikart G (1996) Description and power analysis of two tests for detecting recent population bottlenecks from allele frequency data. Genetics 144:2001–2014
Cornuet JM, Santos F, Beaumont MA et al (2008) Inferring population history with DIY ABC: a user-friendly approach to approximate Bayesian computation. Bioinformatics 24:2713–2719
Cornuet JM, Ravigne V, Estoup A (2010) Inference on population history and model checking using DNA sequence and microsatellite data with the software DIYABC (v1.0). BMC Bioinform 11:1
Dalmon A, Halkett F, Granier M et al (2008) Genetic structure of the invasive pest Bemisia tabaci: evidence of limited but persistent genetic differentiation in glasshouse populations. Heredity 100:316–325
Delmotte F, Leterme N, Gauthier JP et al (2002) Genetic architecture of sexual and asexual populations of the aphid Rhopalosiphum padi based on allozyme and microsatellite markers. Mol Ecol 11:711–723
Delmotte F, Sabater-Munoz B, Prunier-Leterme N et al (2003) Phylogenetic evidence for hybrid origins of asexual lineages in an aphid species. Evolution 57:1291–1303
Desneux N, Stary P, Delebecque CJ et al (2009) Cryptic species of parasitoids attacking the soybean aphid (Hemiptera: Aphididae) in Asia: Binodoxys communis and Binodoxys koreanus (Hymenoptera: Braconidae: Aphidiinae). Ann Entomol Soc Am 102:925–936
Dorken ME, Eckert CG (2001) Severely reduced sexual reproduction in northern populations of a clonal plant, Decodon verticillatus (Lythraceae). J Ecol 89:339–350
Estoup A, Guillemaud T (2010) Reconstructing routes of invasion using genetic data: why, how and so what? Mol Ecol 19:4113–4130
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14:2611–2620
Excoffier L, Smouse PE, Quattro JM (1992) Analysis of molecular variance inferred from metric distances among DNA haplotypes—application to human mitochondrial DNA restriction data. Genetics 131:479–491
Excoffier L, Laval G, Schneider S (2005) Arlequin (version 3.0): an integrated software package for population genetics data analysis. Evol Bioinform 1:47–50
Fonseca DM, Widdel AK, Hutchinson M et al (2010) Fine-scale spatial and temporal population genetics of Aedes japonicus, a new US mosquito, reveal multiple introductions. Mol Ecol 19:1559–1572
Gauffre B, Coeur d’acier A (2006) New polymorphic microsatellite loci, cross-species amplification and PCR multiplexing in the black aphid, Aphis fabae Scopoli. Mol Ecol Notes 6:440–442
Guillemaud T, Beaumont MA, Ciosi M et al (2010) Inferring introduction routes of invasive species using approximate Bayesian computation on microsatellite data. Heredity 104:88–99
Harrison JS, Mondor EB (2011) Evidence for an invasive aphid ‘‘superclone’’: extremely low genetic diversity in oleander aphid (Aphis nerii) populations in the southern United States. PLoS One 6:1–6
Heimpel GE, Frelich LE, Landis DA et al (2010) European buckthorn and Asian soybean aphid as components of an extensive invasional meltdown in North America. Biol Invasions 12:2913–2931
Hill CB, Kim KS, Crull L et al (2009) Inheritance of resistance to the soybean aphid in soybean PI 200538. Crop Sci 49:1193–1200
Hill CB, Crull L, Herman TK et al (2010) A new soybean aphid (Hemiptera: Aphididae) biotype identified. J Econ Entomol 103:509–515
Kanbe T, Akimoto SI (2009) Allelic and genotypic diversity in long-term asexual populations of the pea aphid, Acyrthosiphon pisum in comparison with sexual populations. Mol Ecol 18:801–816
Kim KS, Hill CB, Hartman GL et al (2008) Discovery of soybean aphid biotypes. Crop Sci 48:923–928
Kim H, Hoelmer KA, Lee W et al (2010) Molecular and morphological identification of the soybean aphid and other Aphis species on the primary host Rhamnus davurica in Asia. Ann Entomol Soc Am 103:532–543
Kim Y, Kim M, Hong KJ et al (2011) Outbreak of an exotic flatid, Metcalfa pruinosa (Say) (Hemiptera: Flatidae), in the capital region of Korea. J Asia-Pac Entomol 14:473–478
Lavandero B, Figueroa CC, Ramirez CC et al (2009) Isolation and characterization of microsatellite loci from the woolly apple aphid Eriosoma lanigerum (Hemiptera: Aphididae: Eriosomatinae). Mol Ecol Resour 9:302–304
Llewellyn KS, Loxdale HD, Harrington R et al (2003) Migration and genetic structure of the grain aphid (Sitobion avenae) in Britain related to climate and clonal fluctuation as revealed using microsatellites. Mol Ecol 12:21–34
Lozier JD, Roderick GK, Mills NJ (2007) Genetic evidence from mitochondrial, nuclear, and endosymbiont markers for the evolution of host plant associated species in the aphid genus Hyalopterus (Hemiptera: Aphididae). Evolution 61:1353–1367
Lozier JD, Roderick GK, Mills NJ (2009) Tracing the invasion history of mealy plum aphid, Hyalopterus pruni (Hemiptera: Aphididae), in North America: a population genetics approach. Biol Invasions 11:299–314
Margaritopoulos JT, Kasprowicz L, Malloch GL et al (2009) Tracking the global dispersal of a cosmopolitan insect pest, the peach potato aphid. BMC Ecol 9:13
Michel AP, Zhang W, Jung JK et al (2009a) Cross-species amplification and polymorphism of microsatellite loci in the soybean aphid, Aphis glycines. J Econ Entomol 102:1389–1392
Michel AP, Zhang W, Jung JK et al (2009b) Population genetic structure of Aphis glycines. Environ Entomol 38:1301–1311
Michel AP, Zhang W, Mian MAR (2010) Genetic diversity and differentiation among laboratory and field populations of the soybean aphid, Aphis glycines. B Entomol Res 100:727–734
Miller N, Estoup A, Toepfer S et al (2005) Multiple transatlantic introductions of the western corn rootworm. Science 310:992
Orantes LC, Zhang W, Mian MAR et al (2012) Maintaining genetic diversity and population panmixia through dispersal and not gene flow in a holocyclic heteroecious aphid species. Heredity 109:127–134
Oosterhout CV, Hutchinson WF, Wills DPM et al (2004) Micro-checker: software for identifying and correcting genotyping errors in microsatellite data. Mol Ecol Notes 4:535–538
Paetkau D, Slade R, Burden M et al (2004) Genetic assignment methods for the direct, real-time estimation of migration rate: a simulation-based exploration of accuracy and power. Mol Ecol 13:55–65
Peccoud J, Figueroa CC, Silva AX et al (2008) Host range expansion of an introduced insect pest through multiple colonizations of specialized clones. Mol Ecol 17:4608–4618
Peccoud J, Ollivier A, Plantegenest M et al (2009) A continuum of genetic divergence from sympatric host races to species in the pea aphid complex. Proc Natl Acad Sci USA 106:7495–7500
Piry S, Luikart G, Cornuet JM (1999) BOTTLENECK: a computer program for detecting recent reductions in the effective population size using allele frequency data. J Hered 90:502–503
Piry S, Alapetite A, Cornuet JM et al (2004) GENECLASS2: a software for genetic assignment and first-generation migrant detection. J Hered 95:536–539
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Ragsdale DW, Voegtlin DJ, O’Neil RJ (2004) Soybean aphid biology in North America. Ann Entomol Soc Am 97:204–208
Ragsdale DW, Landis DA, Brodeur J et al (2011) Ecology and management of the soybean aphid in North America. Annu Rev Entomol 56(56):375–399
Rannala B, Mountain JL (1997) Detecting immigration by using multilocus genotypes. Proc Natl Acad Sci USA 94:9197–9201
Raymond M, Rousset F (1995) Genepop (version-1.2)—population genetics software for exact tests and ecumenicism. J Hered 86:248–249
Rice WR (1989) Analyzing tables of statistical tests. Evolution 43:223–225
Sax DF, Stachowicz JJ, Gaines SD (2005) Species invasions: insights into ecology, evolution, and biogeography. Sinauer Associates, Sunderland
Torchin ME, Lafferty KD, Dobson AP et al (2003) Introduced species and their missing parasites. Nature 421:628–630
Vanlerberghe-Masutti F, Chavigny P, Fuller SJ (1999) Characterization of microsatellite loci in the aphid species Aphis gossypii Glover. Mol Ecol 8:693–695
Venette RC, Ragsdale DW (2004) Assessing the invasion by soybean aphid (Homoptera: Aphididae): where will it end? Ann Entomol Soc Am 97:219–226
Weir BS, Cockerham CC (1984) Estimating F-statistics for the analysis of population structure. Evolution 38:1358–1370
Wilson GA, Rannala B (2003) Bayesian inference of recent migration rates using multilocus genotypes. Genetics 163:1177–1191
Wilson ACC, Sunnucks P, Hales DF (2003) Heritable genetic variation and potential for adaptive evolution in asexual aphids (Aphidoidea). Biol J Linn Soc 79:115–135
Wilson ACC, Massonnet B, Simon JC et al (2004) Cross-species amplification of microsatellite loci in aphids: assessment and application. Mol Ecol Notes 4:104–109
Wu ZS, Schenk-Hamlin D, Zhan WY et al (2004) The soybean aphid in China: a historical review. Ann Entomol Soc Am 97:209–218
Yao I, Akimoto SI, Hasegawa E (2003) Isolation of microsatellite markers from the drepanosiphid aphid Tuberculatus quercicola (Homoptera, Aphididae). Mol Ecol Notes 3:542–543
Yencho GC, Cohen MB, Byrne PF (2000) Applications of tagging and mapping insect resistance loci in plants. Annu Rev Entomol 45:393–422
Zepeda-Paulo FA, Simon JC, Ramirez CC et al (2010) The invasion route for an insect pest species: the tobacco aphid in the New World. Mol Ecol 19:4738–4752
Zhu GP, Bu WJ, Gao YB et al (2012) Potential geographic distribution of brown marmorated stink bug invasion (Halyomorpha halys). PLoS One 7:e31246
Zygouridis NE, Augustinos AA, Zalom FG et al (2009) Analysis of olive fly invasion in California based on microsatellite markers. Heredity 102:402–412
Acknowledgments
This study was supported by USDA-ARS Specific Cooperative Agreement no. 58-1926-7-154F. It was also supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education, Science and Technology (2011-0012300). I appreciate all scientists who helped us collect the regional samples of the soybean aphid (See Table 1).
Author information
Authors and Affiliations
Corresponding authors
Electronic supplementary material
Below is the link to the electronic supplementary material.
Fig. S1
Plots output by DIYABC showing the posterior probability (y-axis) of the three scenarios through the direct (left) and the logistic (right) approaches as output by DIYABC. The x-axis correspond to the different n δ values used in the computations. On upper row, results have been obtained by performing the first analysis with three scenarios (A1, A2, A3). On bottom row, results have been obtained by performing the first analysis with three scenarios (B1, B2, B3). (JPEG 464 kb)
Rights and permissions
About this article

{kind=link}
Cite this article
Kim, H., Hoelmer, K.A. & Lee, S. Population genetics of the soybean aphid in North America and East Asia: test for introduction between native and introduced populations. Biol Invasions 19, 597–614 (2017). https://doi.org/10.1007/s10530-016-1299-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10530-016-1299-7