
Abstract
Objective
This study is to explore the exact roles of extracellular vesicle (EVs) miRNAs from brain microvascular pericytes in the pathogenesis of hypertension.
Results
Forty-eight significantly differentially expressed miRNAs were identified, of which 17 were found to be upregulated and 31 were found to be downregulated in brain microvascular pericytes of spontaneous hypertensive rats, compared with that of normotension Wistar Kyoto rats. The GO enrichment analysis verified that the target genes were enriched in signaling pathways and molecular functions, such as metal ion binding, nucleotide binding and ATP binding. The KEGG analysis indicated that the target genes were enriched in Linoleic acid, alpha-linolenic acid and sphingolipid metabolism pathways.
Conclusions
Several EV derived miRNAs, such as miR-21-5p, let-7c-5p and let-7a-5p, were found to be abnormally expressed in brain microvascular pericytes obtained from spontaneous hypertensive rats, compared with that of normotension Wistar Kyoto rats. The results of our research provide more insights into the functional link between brain microvascular pericytes and the pathogenesis of hypertension.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Hypertension is a complex disease and its pathogenesis is determined by multiple genetic and environmental factors (Carrick et al. 2018). Hypertension is a major risk factor for heart disease, myocardial infarction, vascular disease, stroke and chronic kidney disease, as proved by genetic and epidemiological studies (Serne et al. 2007). Pericytes, which are mural cells that wrap around endothelial cells, have drawn attention as regulators of vascular morphogenesis (Ferland-McCollough et al. 2017). Previous studies have shown that pericytes, which perform critical functions throughout the body, have been implicated in various processes, including the development of cardiovascular and cerebrovascular diseases, during recent years and therefore may be potential new targets for antiangiogenic therapies (Armulik et al. 2011; Ferland-McCollough et al. 2017). For example, our previous study identified several differentially expressed genes and signaling pathways in brain microvascular pericytes between SHR (spontaneous hypertensive rats) and WKY (normotension Wistar Kyoto) rats through transcriptome profile analysis (Yuan et al. 2018). We also detected the m6A methylation levels in WKY pericytes and SHR pericytes via m6A high throughput sequencing. Our previous study revealed the respective m6A landscapes and identified an epitranscriptomic mechanism involved in the development of mammalian hypertension (Wu et al. 2019).
microRNAs (miRNAs) are small endogenous non coding RNAs with a length of 18–25 nucleotides that bind to the 3′ or 5′ untranslated regions (UTR) of genes, which correspondingly regulate their expression at a post-transcriptional level (Garcia-Contreras et al. 2017). miRNAs exert their effects on cells that they are synthesized in, and are also released into the extracellular matrix and are transported in body fluids, such as blood and urine (Hannafon et al. 2016). Increasing evidence has proved that miRNAs play significant roles in multiple cellular and molecular events, and that their dysregulation has been identified during the pathogenesis of various diseases, including hypertension (Ebrahimkhani et al. 2017).
Extracellular vesicles (EVs) are released from most eukaryotic cells and contain lipids, proteins, mRNAs and miRNAs (Sugimachi et al. 2015). EVs derived from pericytes can be easily taken up by endothelial cells, which are another important component of the neurovascular unit (Gaceb et al. 2018). Studies on pericyte EVs in spinal cord injury (SCI) found that they could not only improve endothelial ability to regulate blood flow, but also form a protective barrier around spinal cord microvascular endothelial cells (Yuan et al. 2019). Recent research has proved that miRNAs can be transported in body fluids while within EVs (Hornick et al. 2015). Once the miRNAs are released into the extracellular fluid, it was found that EVs fuse with other cells and transfer the miRNAs to acceptor cells (Bhome et al. 2018). Exosomal miRNAs may play critical roles in cell communication and can be used as disease detection and monitoring biomarkers (Zhang et al. 2015). Exosomal miRNA signatures have recently been identified in several cancers, including gastric cancer (Ren et al. 2019), colorectal cancer (Tang et al. 2019), acute myeloid leukemia (Hornick et al. 2015) and hepatocellular carcinoma (Sugimachi et al. 2015). However, EV miRNAs in pericytes have not been well analyzed for their hypertension diagnosis or monitoring abilities.
In this study, we explored the differential expression patterns of EVs in brain microvascular pericytes from SHR and WKY rats using miRNA profiling analysis. Several EV miRNAs in brain microvascular pericytes were identified as potential biomarkers or potential therapeutic targets for hypertension. Collectively, our findings further elucidated the molecular mechanisms underlying the development of hypertension.
Materials and methods
Sample preparation and identification
Experiments on laboratory animals were authorized by the Laboratory Animal Care and Ethics Committee of the Institute of Microcirculation, Peking Union Medical College & Chinese Academy of Medical Sciences. WKY rats (n = 10) and SHR rats (n = 10) were purchased from Vital River Laboratory Animal Technology Co. Ltd. (license No. SCXK 2016-0006, Beijing, China). The brains of the rats were immersed in an ice-cold isolation buffer immediately after decapitation. After the tissues were removed, micro-vessels were isolated, as previously described (Yuan et al. 2018). When the pericytes reached 60–80% confluency, they were cultured in an EV depleted FBS-containing medium (EXO-FBS-250 A-1; System Biosciences, Mountain View, CA, United States) for an additional 48 h. Then, the EVs were isolated from the samples using multi-step centrifugation, as previously reported (Villarroya-Beltri et al. 2013; Xin et al. 2012). In brief, the supernatant collected from the cultured pericytes were centrifuged at 2000×g for 30 min to remove large debris and dead cells, followed by centrifugation at 10,000×g for 30 min to remove small cell debris and finally at 100,000×g for 70 min at 4 °C. Thereafter, contaminating proteins were removed through centrifugation at 100,000×g for 70 min at 4 °C. (Beckman XPN-100, SW28Ti Rotor). The purified EVs were resuspended in PBS for functional assays and protein detection. The protein concentrations of the EVs were determined using a BCA Protein Assay Kit (Thermo Fisher Scientific).
Sample identification
Transmission electron microscopy (TEM)
The morphology of the EVs obtained from the pericytes was observed using TEM. EV pellets were fixed in 2% paraformaldehyde (PFA)-cacodylate buffer and were loaded onto copper grids covered with formvar for 20 min. Then, the EVs were fixed in 1% (w/v) glutaraldehyde for 5 min. The grids were washed and contrasted using 4% uranyl acetate for 5 min, dried, and then observed under TEM (FEI TECNAI G2, 120 kV).
NTA: size distribution analysis of pericyte EVs
The suspensions with vesicles were analyzed using a Nano-Sight LM10 instrument (Malvern, Worcestershire, United Kingdom). A monochromatic laser beam was used to illuminate the diluted samples at 405 nm to record a 60 s video at a mean frame rate of 25 frames/s. The EV samples were analyzed using NTA software (version 3.0, Nano-Sight) to first distinguish and then follow each particle on an optimal frame-by-frame basis to track Brownian movement and measure the same from frame to frame. The size of the particles were determined using the two-dimensional Stokes–Einstein equation, based on the velocity of particle movement. The mean, mode and median EV size in each video was used to calculate sample concentration expressed as nanoparticles/mL.
Western blotting analysis
The EV samples stored in a refrigerator at – 80 °C were added into the loading buffer. The protein was denatured by heating at 99 °C for 5 min. The protein samples were added into the sample hole and protein electrophoresis was performed on SDS-PAGE at a voltage of 80 V. After the sample was compressed into a straight line, electrophoresis was continued at a voltage of 100 V. Thereafter, the PVDF membrane (Millipore) was kept at 100 V at a constant pressure for 2 h, and 5% skim milk powder was used for sealing for 40 min, and was incubated with a primary antibody (CD9, ab92726, 1:2000; CD81, ab109201, 1:1000; ABCAM, MA, USA) overnight in a shaking bed at 4 °C. TBST buffer was used to wash the PVDF membrane, which was then incubated at room temperature with the secondary antibody (Goat Anti-Rabbit IgG H&L (HRP) (ab6721), 1:3000) within 1 h for 5 min each time and repeated 3 times. Then, TBST was again used to wash the membrane for 10 min and was repeated 3 times. Then, a luminescent solution was added and exposure was performed using the automatic developer.
RNA extraction
The EV RNA was extracted using a RNeasy maxi kit (QIAGEN, GER) and was kept in RNase free water. A NanoPhotometer® Spectrophotometer (IMPLEN, CA, USA) was used to measure RNA purity. A Qubit RNA Assay Kit and Qubit 2.0 Flurometer (Life Technologies, CA, USA) were used to determine RNA concentration. The RNA Nano 6000 Assay Kit of the Agilent Bioanalyzer 2100 system (Agilent Technologies, CA, USA) was used to assess RNA integrity.
Preparation of the sequencing library and small RNA sequencing
A NEBNext Multiplex Small RNA Library Prep Set for Illumina (NEB, USA.) was used to generate the sequencing libraries, following the manufacturer’s instructions. In order to attribute sequences to each sample, the index codes were also added. DNA fragments with a length of 140–160 bp were recovered and dissolved in 8 μL of elution buffer. Finally, DNA High Sensitivity Chips were used to access library quality on the Agilent Bioanalyzer 2100 system. After clustering the index-coded samples, the cDNA library was sequenced on an Illumina Hiseq 2500/2000 high-throughput sequencing platform, with 50 bp long single-end reads being generated.
Processing and assembly of sequencing raw data
Clean reads were obtained by removing reads containing ploy-N or 5′ adapter contaminants, not containing 3′ adapter or the insert tag, or containing ploy A, T, G or C, while low-quality reads were also removed from raw data. We used Bowtie software (Langmead et al. 2009) to map clean reads on the reference genome. Known miRNAs were identified and mapped using small RNA tags, with miRBase 20.0 used as the reference. Then, small RNA reads were mapped on the RepeatMasker, Rfam database to exclude reads originating from protein-coding genes, repeat sequences, rRNA, tRNA, snRNA and snoRNA.
Differentially expressed miRNAs
miRNAs were quantified as TPM (transcript per million), which was calculated using the formula: normalized expression = unique mapped read count/Total reads × 100,0000. DESeq of R software (1.8.4) was used to identify differentially expressed miRNAs between two conditions. Significantly differentially expressed miRNAs were defined as having an adjusted p value of < 0.05 and fold change of > 2. The target genes of the miRNAs were further predicted using miRanda software (Enright et al. 2003).
GO and KEGG enrichment analysis
Gene ontology (GO) term analysis was applied to target genes with significantly differentially expressed miRNAs between EVs from brain microvascular pericytes of SHR and WKY, using GOseq, which adjusted gene length bias. Kyoto Encyclopedia of Genes and Genomes (KEGG) is a database used for the systematic analysis of gene functions and genomic information, which has been designed to understand the functions of large-scale molecular datasets generated by high-throughput sequencing (https://www.genome.jp/kegg/). KOBAS software (Mao et al. 2005) was used to identify the significant enrichment pathways of the target genes.
Results
Characterization of pericyte EVs
We analyzed the miRNA profile of the EVs isolated from the medium of the brain microvascular pericytes of ten spontaneous hypertensive rats (SHR) and 10 normotension Wistar Kyoto rats (WKY). Figure 1 illustrates the workflow of this study.

The experimental procedure of the whole research
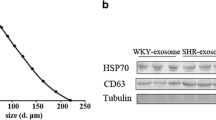
EVs isolated from brain microvascular pericytes of SHR and WKY rats were morphologically confirmed using TEM (Fig. 2a). The EVs extracted through hypervelocity centrifugation showed an obvious saucer like double-layer membrane structure under an electron microscope, and the particle diameter was approximately 100 nm. In addition, the diameter and size distribution of the pericyte EVs were further analyzed using NTA (Fig. 2b). Particle diameter showed a distribution from 100 to 350 nm in width, with a mean width of 266 nm. Two EV markers, CD9 and CD81, were clearly detected using western blotting analysis, which further confirmed the identity of the isolated EVs (Fig. 2c, d).

Characterization of brain microvascular pericytes EVs from WKY and SHR. a Transmission electron microscopy shows the pericytes EVs. The scale bar represents 200 nm. b The diagram shows the size and concentration of EVs derived from pericytes by use of Nanoparticle Tracking Analysis (NTA). c, d Western blot identifies pericytes EVs by detecting EVs marker CD9 (c) and CD81 (d)
Small RNA classification
EVs contain numerous types of small RNA, such as miRNA, rRNA, tRNA, snoRNA and snRNA. These small RNAs can be classified by mapping the unique reads to noncoding RNA databases, such as miRBase, RepeatMasker and Rfam. miRNAs of brain microvascular pericyte EVs of WKY and SHR rats contained 72.3% and 78.9% of small RNAs, respectively (Fig. 3a), which included rRNAs (25.02% and 19.58%, respectively), tRNAs (1.24% and 1.19%, respectively), snoRNAs (1.33% and 0.26%, respectively), snRNAs (0.19% and 0.08%, respectively) and other small RNAs (1.06% and 0.18%, respectively). Furthermore, 386 known miRNAs were identified in the WKY rat sample, while 225 known miRNAs were identified in the SHR rat sample, with an overlap of 219 miRNAs between the groups (Fig. 3b).

Small RNA classification in brain microvascular pericytes EVs from WKY and SHR. a The percentage of small RNA categories in WKY (left) and SHR (right). b Venn diagram showing the count of known miRNAs in WKY and SHR pericytes EVs and the overlap count between two groups
Differentially expressed miRNAs
miRNA profiling of the EVs derived from brain microvascular pericytes identified 48 significantly differentially expressed miRNAs, of which 17, including let-7c-5p, let-7a-5p, miR-11980, miR-122-5p and miR-378a-3p, were upregulated, while 31, including miR-6240, miR-26a-5p, let-7i-5p and miR-1285, were downregulated in SHR rats, compared with that of normotension WKY rats (Table 1). The differentially expressed miRNAs were hierarchically clustered (Fig. 4).

Heatmap of differential miRNA expression in brain microvascular pericytes EVs between WKY and SHR
miRNA-gene regulatory network
It is well known that a single miRNA can regulate hundreds of genes, while a single gene can have numerous miRNA binding sites. In order to investigate the influence of miRNAs of brain microvascular pericyte EVs, target genes of the significantly differentially expressed miRNAs were identified using miRanda and TargetScan. The resulting miRNA-gene and gene–gene pairs were constructed using Cytoscape software (version 3.5.0), where edges between the miRNAs and target genes represent a potential regulatory relationship and edges between genes represent an expression correlation (Fig. 5). Interestingly, we found that matrix metalloproteinases (MMPs), including Mmp1, Mmp2, Mmp7, Mmp8, Mmp9 and Mmp16, are involved in the network and that Mmp13, Mmp19 and Vegfa were the central nodes that regulated most of the significantly differentially expressed miRNAs.

The miRNA–gene interaction networks from differentially expressed miRNAs in brain microvascular pericytes EVs. The green circles represent differentially expressed miRNAs and the red rectangles represent target genes of differentially expressed miRNAs
GO analysis of the target genes
In order to further study the tumor biology associated with specific EV-related miRNAs of SHR rats, compared with that of normotension WKY rats, we performed a bioinformatics analysis to identify proteins that are regulated by differentially expressed miRNAs and also to assess their molecular and biological functions.
GO enrichment analysis was performed on the target genes of significantly differentially expressed miRNAs between brain microvascular pericyte EVs of SHR and WKY rats (Fig. 6). The results indicated that the target genes are mainly enriched in biological process (BP), including ‘biological process’, ‘regulation of transcription’, ‘transport’ and ‘signal transduction’, while the cellular components (CC) included ‘membrane’, ‘integral component of membrane’, ‘nucleus’ and ‘cytoplasm’. In addition, molecular functions (MF) included ‘metal ion binding’, ‘molecular function’, ‘nucleotide binding’ and ‘ATP binding’.

Major GO enrichment of target genes of significant differentially expressed miRNAs in brain microvascular pericyte EVs between SHR and WKY
KEGG pathway analysis of the target genes
KEGG pathway analysis was also performed on the target genes of significantly differentially expressed miRNAs between brain microvascular pericyte EVs of SHR and WKY rats (Fig. 7). The results indicated that the target genes are enriched in ‘alpha-Linolenic acid metabolism’, ‘Valine, leucine and isoleucine degradation’, ‘Sphingolipid metabolism’, ‘Betalain biosynthesis’, ‘Ether lipid metabolism’, ‘Geraniol degradation’ and ‘Nicotinate and nicotinamide metabolism’.

Top 20 KEGG pathways regulated by the target genes of significant differentially expressed miRNAs in brain microvascular pericyte EVs between SHR and WKY
Discussion
In an era of precision medicine, it is critical to determine biomarkers and targets for the diagnosis and management of hypertension (Spijkers et al. 2011). miRNAs are short non-coding RNAs that regulate mRNAs in a post-transcriptional manner and its deregulation could lead to metastasis and therapy resistance (Hannafon et al. 2016). EVs, which contain different types of proteins, lipids and miRNAs, can travel throughout the body (Alipoor et al. 2019). Increasing evidence has indicated that exosomal miRNAs are promising biomarkers for clinical diagnosis (Liu et al. 2008, 2019). Our previous study showed that brain microvascular pericytes play an important role in the development of spontaneous hypertension (Yuan et al. 2018). In this study, miRNA expression profiling was performed on EVs from brain microvascular pericytes of SHR and WKY rats.
Several EV miRNAs of brain microvascular pericytes were found to be differentially expressed between SHR and WKY rats and may be potential biomarkers or therapeutic targets for hypertension. miR-21, which is well known for its critical roles in the development of hypertension (Sekar et al. 2017), was found to be upregulated in EVs from brain microvascular pericytes in SHR, compared with that of WKY rats. miR-21 has been identified to perform a positive function in mitochondrial translation, while it has been shown to decrease blood pressure and suppress cardiac hypertrophy in SHR (Cengiz et al. 2015; Li et al. 2016). Recently, Li et al. compared the expression of miRNAs in plasma samples of patients with hypertension with that of healthy controls (Chen et al. 2012). Among the 27 miRNAs, 9 were found to be upregulated and 18 were found to be downregulated, with let-7e being upregulated 1.7 times, compared with that of the control group (p < 0.01). Compared with endothelial progenitor cells (EPCs), the expression level of let-7e in circulating ECs was found to be higher than that of the control group, which is consistent with the expression trend found in plasma, but the change in let-7e level in ECs was opposite to that of plasma. In this study, we found that let-7a and let-7c was upregulated and that let-7i was down-regulated in the SHR group, suggesting that the mechanism by which the let-7 family is involved in hypertension needs to be further explored. These results together support the suggestion that the miRNAs mentioned above in brain microvascular pericytes are potential biomarkers or potential therapeutic targets for hypertension.
GO enrichment analysis of the target genes of significantly differentially expressed miRNAs between brain microvascular pericytes EVs of SHR and WHK rats showed that the genes are enriched in biological processes and molecular functions, which are consistent with the regulatory role of these miRNAs in transcription and translation processes (Liu et al. 2008). Hypertension is characterized by numerous cell membrane abnormalities associated with receptor characteristics, enzymatic activity and transmembrane ion exchange (Spijkers et al. 2011), which are interrelated with cellular signal transduction (Murthy et al. 2017). These previous findings are consistent with our findings, indicating that the target genes are enriched in transport and signal transduction, and molecular functions, such as metal ion binding, nucleotide binding and ATP binding. Alterations in these biological processes and/or molecular functions may play critical roles in the pathological processes involved in the development of hypertension (Majzunova et al. 2013).
In addition, numerous enrichment pathways these target genes are involved in were also identified through KEGG analysis (Shen et al. 2010). Previous reports have revealed that low levels of linoleic acid and alpha-linolenic acid, and high levels of arachidonic acid in plasma phospholipids can decrease the prevalence of hypertension (Tsukamoto and Sugawara 2018). An increasing number of reports have revealed the critical roles of sphingolipids metabolism in the pathogenesis of hypertension (Fenger et al. 2015). A recent study proved that sphingolipids are key mediators of cellular signal transduction, since they are can regulate vascular contractility and growth (Borodzicz et al. 2015). Spijkers et al. further proved that the remarkable transformation in vascular sphingolipid biology is associated with hypertension (Spijkers et al. 2011). Taken together, our results suggest that EVs in brain microvascular pericytes might perform fundamental roles in hypertension via regulating these pathways.
Conclusion
In this study, several EV miRNAs, such as miR-21-5p, let-7c-5p and let-7a-5p, were found to be abnormally expressed in brain microvascular pericyte EVs of SHR, compared with that of WKY rats. GO and KEGG enrichment analysis of the predicted target genes of significantly differentially expressed miRNAs identified hypertension specific genes and signaling pathways. We identified several potential novel biomarkers or therapeutic targets that can be used for the diagnosis and treatment of hypertension, deepening our understanding on the molecular mechanisms involved in hypertension.
Abbreviations
- UTR:
-
Untranslated regions
- EVs:
-
Extracellular vesicles
- SCI:
-
Spinal cord injury
- TEM:
-
Transmission electron microscopy
- NTA:
-
Nanoparticle tracking analysis
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- SHR:
-
Spontaneous hypertensive rats
- WKY:
-
Wistar Kyoto rats
- GO:
-
Gene ontology
- BP:
-
Biology process
- CC:
-
Cellular component
- MF:
-
Molecular function
- SLC7A1:
-
Solute carrier family 7 member 1
References
Alipoor SD et al (2019) Serum exosomal miRNAs are associated with active pulmonary tuberculosis. Dis Markers 2019:1907426. https://doi.org/10.1155/2019/1907426
Armulik A, Genove G, Betsholtz C (2011) Pericytes: developmental, physiological, and pathological perspectives, problems, and promises. Dev Cell 21:193–215. https://doi.org/10.1016/j.devcel.2011.07.001
Bhome R, Del Vecchio F, Lee GH, Bullock MD, Primrose JN, Sayan AE, Mirnezami AH (2018) Exosomal microRNAs (exomiRs): small molecules with a big role in cancer. Cancer Lett 420:228–235. https://doi.org/10.1016/j.canlet.2018.02.002
Borodzicz S, Czarzasta K, Kuch M, Cudnoch-Jedrzejewska A (2015) Sphingolipids in cardiovascular diseases and metabolic disorders. Lipids Health Dis 14:55. https://doi.org/10.1186/s12944-015-0053-y
Carrick D et al (2018) Hypertension, microvascular pathology, and prognosis after an acute myocardial infarction. Hypertension 72:720–730. https://doi.org/10.1161/hypertensionaha.117.10786
Cengiz M et al (2015) Circulating miR-21 and eNOS in subclinical atherosclerosis in patients with hypertension. Clin Exp Hypertension 37:643–649. https://doi.org/10.3109/10641963.2015.1036064
Chen X, Liang H, Zhang J, Zen K, Zhang CY (2012) Secreted microRNAs: a new form of intercellular communication. Trends Cell Biol 22:125–132. https://doi.org/10.1016/j.tcb.2011.12.001
Ebrahimkhani S et al (2017) Exosomal microRNA signatures in multiple sclerosis reflect disease status. Sci Rep 7:14293. https://doi.org/10.1038/s41598-017-14301-3
Enright AJ, John B, Gaul U, Tuschl T, Sander C, Marks DS (2003) MicroRNA targets in Drosophila. Genome Biol 5:R1. https://doi.org/10.1186/gb-2003-5-1-r1
Fenger M, Linneberg A, Jeppesen J (2015) Network-based analysis of the sphingolipid metabolism in hypertension. Front Genet 6:84. https://doi.org/10.3389/fgene.2015.00084
Ferland-McCollough D, Slater S, Richard J, Reni C, Mangialardi G (2017) Pericytes, an overlooked player in vascular pathobiology. Pharmacol Ther 171:30–42. https://doi.org/10.1016/j.pharmthera.2016.11.008
Gaceb A, Barbariga M, Ozen I, Paul G (2018) The pericyte secretome: potential impact on regeneration. Biochimie 155:16–25. https://doi.org/10.1016/j.biochi.2018.04.015
Garcia-Contreras M, Shah SH, Tamayo A, Robbins PD, Golberg RB, Mendez AJ, Ricordi C (2017) Plasma-derived exosome characterization reveals a distinct microRNA signature in long duration type 1 diabetes. Sci Rep 7:5998. https://doi.org/10.1038/s41598-017-05787-y
Hannafon BN et al (2016) Plasma exosome microRNAs are indicative of breast cancer. Breast Cancer Res 18:90. https://doi.org/10.1186/s13058-016-0753-x
Hornick NI, Huan J, Doron B, Goloviznina NA, Lapidus J, Chang BH, Kurre P (2015) Serum exosome MicroRNA as a minimally-invasive early biomarker of AML. Sci Rep 5:11295. https://doi.org/10.1038/srep11295
Langmead B, Trapnell C, Pop M, Salzberg SL (2009) Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10:R25. https://doi.org/10.1186/gb-2009-10-3-r25
Li H et al (2016) MicroRNA-21 Lowers blood pressure in spontaneous hypertensive rats by upregulating mitochondrial translation. Circulation 134:734–751. https://doi.org/10.1161/circulationaha.116.023926
Liu X, Fortin K, Mourelatos Z (2008) MicroRNAs: biogenesis and molecular functions. Brain Pathol 18:113–121. https://doi.org/10.1111/j.1750-3639.2007.00121.x
Liu X, Yuan W, Yang L, Li J, Cai J (2019) miRNA profiling of exosomes from spontaneous hypertensive rats using next-generation sequencing. J Cardiovasc Trans Res 12:75–83. https://doi.org/10.1007/s12265-017-9784-7
Majzunova M, Dovinova I, Barancik M, Chan JY (2013) Redox signaling in pathophysiology of hypertension. J Biomed Sci 20:69. https://doi.org/10.1186/1423-0127-20-69
Mao X, Cai T, Olyarchuk JG, Wei L (2005) Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics 21:3787–3793. https://doi.org/10.1093/bioinformatics/bti430
Murthy M, Kurz T, O'Shaughnessy KM (2017) WNK signalling pathways in blood pressure regulation. Cell Mol Life Sci 74:1261–1280. https://doi.org/10.1007/s00018-016-2402-z
Ren W et al (2019) Exosomal miRNA-107 induces myeloid-derived suppressor cell expansion in gastric cancer. Cancer Manage Res 11:4023–4040. https://doi.org/10.2147/cmar.S198886
Sekar D, Shilpa BR, Das AJ (2017) Relevance of microRNA 21 in different types of hypertension. Curr Hypertens Rep 19:57. https://doi.org/10.1007/s11906-017-0752-z
Serne EH, de Jongh RT, Eringa EC, Ijzerman RG, Stehouwer CD (2007) Microvascular dysfunction: a potential pathophysiological role in the metabolic syndrome. Hypertension 50:204–211. https://doi.org/10.1161/hypertensionaha.107.089680
Shen E, Diao X, Wei C, Wu Z, Zhang L, Hu B (2010) MicroRNAs target gene and signaling pathway by bioinformatics analysis in the cardiac hypertrophy. Biochem Biophys Res Commun 397:380–385. https://doi.org/10.1016/j.bbrc.2010.05.116
Spijkers LJ et al (2011) Hypertension is associated with marked alterations in sphingolipid biology: a potential role for ceramide. PLoS ONE 6:e21817. https://doi.org/10.1371/journal.pone.0021817
Sugimachi K et al (2015) Identification of a bona fide microRNA biomarker in serum exosomes that predicts hepatocellular carcinoma recurrence after liver transplantation. Br J Cancer 112:532–538. https://doi.org/10.1038/bjc.2014.621
Tang Y, Zhao Y, Song X, Song X, Niu L, Xie L (2019) Tumor-derived exosomal miRNA-320d as a biomarker for metastatic colorectal cancer. J Clin Lab Anal 33:e23004. https://doi.org/10.1002/jcla.23004
Tsukamoto I, Sugawara S (2018) Low levels of linoleic acid and alpha-linolenic acid and high levels of arachidonic acid in plasma phospholipids are associated with hypertension. Biomed Reps 8:69–76. https://doi.org/10.3892/br.2017.1015
Villarroya-Beltri C, Gutierrez-Vazquez C, Sanchez-Madrid F, Mittelbrunn M (2013) Analysis of microRNA and protein transfer by exosomes during an immune synapse. Methods Mol Biol 1024:41–51. https://doi.org/10.1007/978-1-62703-453-1_4
Wu Q, Yuan X, Han R, Zhang H, Xiu R (2019) Epitranscriptomic mechanisms of N6-methyladenosine methylation regulating mammalian hypertension development by determined spontaneously hypertensive rats pericytes. Epigenomics. https://doi.org/10.2217/epi-2019-0148
Xin H et al (2012) Exosome-mediated transfer of miR-133b from multipotent mesenchymal stromal cells to neural cells contributes to neurite outgrowth. Stem Cells 30:1556–1564. https://doi.org/10.1002/stem.1129
Yuan X, Wu Q, Liu X, Zhang H, Xiu R (2018) Transcriptomic profile analysis of brain microvascular pericytes in spontaneously hypertensive rats by RNA-Seq. Am J Transl Res 10:2372–2386
Yuan X et al (2019) Exosomes derived from pericytes improve microcirculation and protect blood-spinal cord barrier after spinal cord injury in mice. Front Neurosci 13:319. https://doi.org/10.3389/fnins.2019.00319
Zhang J, Li S, Li L, Li M, Guo C, Yao J, Mi S (2015) Exosome and exosomal microRNA: trafficking, sorting, and function. Genomics Proteomics Bioinf 13:17–24. https://doi.org/10.1016/j.gpb.2015.02.001
Acknowledgements
This study was supported by the innovation fund of the Chinese Academy of Medical Sciences and Peking Union Medical College (Grant Nos. 3332014006 and 3332015123), the CAMS Initiative for Innovative Medicine (CAMS-I2M) (Grant No. 2016-I2M-3-006) and National Natural Science Foundation of China (Grant No. 81801433).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors have no conflicts of interest to declare.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Wu, Q., Yuan, X., Li, B. et al. Differential miRNA expression analysis of extracellular vesicles from brain microvascular pericytes in spontaneous hypertensive rats. Biotechnol Lett 42, 389–401 (2020). https://doi.org/10.1007/s10529-019-02788-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10529-019-02788-x