
Abstract
Objective
To determine the effect of large genomic region deletion in a Saccharomyces cerevisiae strain on tyrosine yield and to identify new genetic modification targets through transcriptome analysis.
Results
TAL was used to produce p-coumaric acid (p-CA) from tyrosine to quantity tyrosine yield. S. cerevisiae mutant strain NK14 with deletion of a 23.8 kb genomic region was identified to have p-CA production of 10.3 mg L− 1, while the wild-type strain BY4741 had p-CA production of 1.06 mg L− 1. Analysis of growth patterns and stress tolerance showed that the deletion did not affect the growth phenotype of NK14. Transcriptome analysis suggested that, compared to BY4741, genes related to glycolysis (ENO2, TKL1) and the tyrosine pathway (ARO1, ARO2, ARO4, ARO7, TYR1) were upregulated in NK14 at different levels. Besides genes related to the tyrosine biosynthetic pathway, amino acid transporters (AVT6, VBA5, THI72) and transcription factor (ARO80) also showed changes in transcription levels.
Conclusions
We developed a strain with improved tyrosine yield and identified new genetic modification candidates for tyrosine production.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Saccharomyces cerevisiae is widely used as a microbial host in the production of various chemicals and pharmaceuticals (Hong and Nielsen 2012). The aromatic amino acid l-tyrosine is an important precursor for the biosynthesis of valuable plant secondary metabolites and serves as a useful research target (Li et al. 2015; Eichenberger et al. 2017; Lehka et al. 2017). Synthesis of tyrosine in yeast starts from two precursors, erythrose-4-phosphate (E4P) and phosphoenolpyruvate (PEP), derived from the pentose phosphate pathway (PPP) and glycolysis, respectively. DAHP synthase (ARO3 and ARO4) catalyzes the first step in the shikimic acid pathway, which involves condensation of PEP and E4P to form 3-deoxy-d-arabino-heptulosonate-7-phosphate (DAHP). ARO1 enzyme then catalyzes five reactions that lead from DAHP to 5-enolpyruvyl-3-shikimate phosphate (EPSP). The last step of the shikimic acid pathway is catalyzed by chorismate synthase ARO2, which converts EPSP into chorismate. Further downstream, from chorismate towards the tyrosine and phenylalanine branch, ARO7 catalyzes the conversion of chorismate to prephenate. The next step is synthesis of phenylpyruvate (PPY), catalyzed by prephenate dehydratase (PHA2), and synthesis of 4-hydroxyphenylpyruvate (HPP), catalyzed by prephenate dehydrogenase (TYR1). Finally, aromatic amino transferases ARO8 and ARO9 can reversibly convert HPP to l-tyrosine or PPY to l-phenylalanine.
Efforts made to build platform strains engineered for tyrosine production have included increasing the precursor supply for the tyrosine pathway via deletion of ZWF1 and overexpression of TAL1 and ENO2 (Deaner and Alper 2017; Mao et al. 2017). Shikimate pathway engineering is also an option via overexpression of mutant ARO4 (Luttik et al. 2008; Koopman et al. 2012; Galanie et al. 2015) or replacement of ARO1 with AroL from Escherichia coli (Rodriguez et al. 2017). Another common method is to remove the competitive pathway by deleting ARO10, PDC5, and PDC6 (Gold et al. 2015; Rodriguez et al. 2017).
In this study, we performed large genomic region (LGR) deletion and obtained mutant strain NK14 with p-CA production of 10.3 mg L− 1, which is 9.8-fold higher than that obtained with the BY4741 strain. We conducted a comparative analysis of the NK14 and BY4741 transcriptomes in an attempt to identify genes and pathways related to tyrosine production.
Materials and methods
Yeast strains, plasmids, and culture conditions
The yeast strains used in this study are listed in Table 1. All the strains with genomic regions deleted and the ARO80 deletion strain were derived from the wild-type BY4741 strain (MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0). Single-gene knockout strains NK141 to NK1412 were derived from strain BY4742 (MATα his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0). Both parental strains were purchased from Euroscarf (Frankfurt, Germany). The plasmids used are shown in Supplementary Table 1. Yeast strains were cultured at 30 °C and 220 rpm. YPD-rich medium was used for growth curve measurements, and synthetic dropout medium (SC-his-tyr) was used for tyrosine fermentation. E. coli DH5α used for plasmid propagation was grown in LB medium at 37 °C and 200 rpm.
Construction of mutant strains
Strains NK141-NK1412 were a gift from Professor Yingjin Yuan (Tianjin University, China) and were constructed via homologous recombination by replacing the ORFs with a kana box (Giaever et al. 2002). Supplementary Table 2 shows details for the strains with single-gene deletion. The ARO80 deletion strain and the mutant strains with deletion of chromosomal region(s) were constructed using the mazF-mediated deletion system previously reported (Liu et al. 2014). The primers used in this study are listed in Supplementary Table 3.
Tyrosine fermentation and HPLC analysis
Plasmid pLC-m7 was transformed in the mutant strains and selected on SC-his plates according to the selection marker. Three colonies from independent transformants were inoculated into fresh medium and incubated overnight. Precultures were inoculated to an OD600 of 0.1 in 250 mL flasks with 50 mL of SC-his-tyr medium. At 24 h time intervals, 1 mL samples were manually collected and centrifuged (500×g, 5 min, room temperature). The supernatant was stored at − 20 °C for p-CA quantification by HPLC.
p-CA was quantified on a HPLC system equipped with an Eclipse XDB-C18 column (4.6 × 150 mm, 5 µm, Agilent) (Gold et al. 2015) using 10 µL aliquots of the culture supernatants. A standard curve was prepared using p-CA standards.
Growth analysis
The growth of strain NK14 was monitored to produce growth curve measurements under normal and stress conditions. A loop full of cells from agar plates was taken to inoculate NK14 and wild-type BY4741 into 3 mL of YPD medium for culture overnight. The cultures were diluted to an optical density at 600 nm (OD600) of 0.1 and then cultured in 15 mL YPD medium in 100 mL flasks. OD600 measurements were performed at 8 h intervals up to 48 h on a microplate reader (PerkinElmer, USA) using a 20 µL culture aliquot mixed with 180 µL of water.
To measure growth under stress conditions, the strains were cultured overnight in liquid YPD medium to the exponential phase and adjusted to OD600 of 1.0 with sterile water. Serial tenfold dilutions of the precultures were spotted onto YPD plates containing 1 M NaCl, 1.5 M sorbitol, or 8% ethanol. To measure the heat shock response, cultures were incubated at 50 °C for 30 min before dilution. Plates were cultured at 30 °C for 3 days and photographed.
Transcriptome analysis
Total RNA was extracted and then enriched using oligo(dT) beads. The enriched mRNA was broken into short fragments to generate cDNA libraries. The cDNA fragments were purified and end-repaired, and then ligated to Illumina sequencing adapters after addition of poly(A). The ligation products were size-selected via agarose gel electrophoresis, amplified via PCR, and then sequenced using an Illumina HiSeq 2500 system (Gene Denovo Biotechnology, Guangzhou, China).
Results
Effects of LGR deletion on p -CA production
Six strains with deletion of genomic regions of different sizes varying from 20 to 77 kb were chosen for tyrosine fermentation. The p-CA production of the mutant strains is shown in Fig. 1. NK14 showed significantly higher p-CA production compared to BY4741 during the whole fermentation period. The fermentation stopping point at 72 h was chosen by considering both p-CA production and nutrient consumption. The p-CA yield for the NK14 strain was 10.3 mg L− 1, which is 9.8-fold higher than that for BY4741. Thus, NK14 could be a good starting point for modification of the tyrosine pathway.

p-CA synthesis of genome region deletion strains. We monitored the tyrosine yield every 24 h during the fermentation through the p-coumaric acid by strains with the TAL plasmid. The strains were cultivated at 30 °C in 50 mL SC-his-tyr medium in flasks. Error bars represent the standard deviation of three biological duplicates
NK14 is a strain with deletion of a genomic region containing 12 genes. To determine whether the improvement in p-CA production is due to deletion of one individual gene, the p-CA production of 12 single-gene knockout strains was measured (Supplementary Fig. 1). Compared to BY4742, which is the starting strain, NK141 and NK148 showed an insignificant improvement in p-CA production. The p-CA production of other strains was also lower than that of the control strain. Hence, the increase in p-CA yield from NK14 cannot be attributed to deletion of an individual gene.
Growth pattern and stress tolerance
LGR deletion in NK14 resulted in an enhanced p-CA yield. To confirm that the strain is a robust p-CA producer, its growth pattern and stress resistance were assessed (Fig. 2). Although strain NK14 carries a deletion of 12 genes, it exhibited a similar growth pattern to BY4741, as shown by the growth curve (Fig. 2a). Furthermore, under high salinity (1 M NaCl), hyperosmotic stress (1.5 M sorbitol), and high ethanol pressure (8% ethanol), both NK14 and wild-type BY4741 showed similar growth (Fig. 2b). NK14 was slightly more sensitive to heat shock than the parental strain.

Growth profile of strain NK14 with enhanced tyrosine production. a Growth curves of strain NK14 and wild type strain BY4741. b Stress tolerance test of strain NK14 and wild type strain BY4741. Serial tenfold dilution of the pre-cultures was spotted onto a solid YPD media incubated at 30°C for 3 days. Inhibitors were 1.5 M sorbitol, 8% ethanol, 1 M NaCl. For heat-shock response, the cell suspension was incubated at 50 °C for 30 min and cooled to 30°C, followed by spotting on YPD plate. The test was repeated three times
Transcriptional response to LGR deletion
Differentially expressed genes (DEGs) were identified using thresholds of FDR < 0.05 and a |log2 fold change (FC)| > 1. This analysis identified 3576 DEGs, of which 1208 were upregulated and 2368 were downregulated. According to the Gene Ontology database, the 3576 DEGs could be classified in three categories: biological processes, cellular components, and molecular functions (Fig. 3a). In the biological process category, DEGs related to cellular, metabolic, and single-organism processes were significantly enriched. In the cellular component class, cell, cell part, and organelle were the top three categories with significant DEG enrichment. For the molecular function category, DEGs were enriched mainly in the binding and catalytic activity classes.

Analysis of DEGs of NK14 based on the GO and KEGG pathways. a GO classification analysis of DEGs: biological process, cellular component, and molecular function. (red) number of DEGs with an enhanced transcription level, (green) number of DEGs with a decreased transcription level. b The top 20 DEG-enriched KEGG pathways. Rich factor: the greater the rich factor is, the greater the enrichment. QValue: indicates the significance level after hypergeometric tests; a smaller value indicates a greater enrichment
We also performed analysis using the KEGG database to determine whether the DEGs were involved in specific pathways. Figure 3b shows the top 20 enriched pathways among all 112 pathways. The significantly enriched pathways were mainly associated with amino acid metabolism (valine, leucine, and isoleucine degradation; beta-alanine metabolism; cysteine and methionine metabolism; valine, leucine, and isoleucine biosynthesis; histidine metabolism; biosynthesis of amino acids) and energy metabolism processes (carbon metabolism; PPP; pyruvate metabolism; glycolysis/gluconeogenesis; fructose and mannose metabolism).
Analysis of DEGs related to tyrosine metabolism
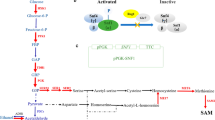
The tyrosine biosynthetic pathway comprises the shikimic acid pathway and l-tyr and l-phe branches. We also focused on the glycolysis pathway and PPP, which provide the precursors E4P and PEP for the tyrosine pathway. We searched for upregulated and downregulated genes involved in these pathways using the SGD database. Figure 4a shows the overall tyrosine biosynthetic pathway from glucose, with upregulated and downregulated genes denoted in red and blue, respectively. The heatmap shows the expression level of genes related to tyrosine biosynthesis (Fig. 4b). Genes involved in the glycolysis pathway were upregulated at different levels. In particular, ENO2 was remarkably upregulated, consistent with the finding that ENO2 overexpression increases the tyrosine yield (Mao et al. 2017). ZWF1 was downregulated, while RKI1 and TKL1 were upregulated in the PPP. For the shikimic acid pathway, higher expression levels of ARO1, ARO2, ARO4, and ARO7 facilitate higher carbon fluxes into the tyrosine pathway.

Transcriptional level of genes included in the tyrosine metabolism. a Simplified map of carbon metabolism leading to the tyrosine pathway in S. cerevisiae. Genes upregulated are labeled in red and downregulated are labeled in blue. G6P glucose-6-phosphate, F6P fructose-6-phosphate, FDP fructose 1,6-bisphosphate, G3P glyceraldehyde-3-phosphate, 1-3BPG 1,3-bisphosphoglycerate, 3PG 3-phosphoglyceric acid, 2PG 2-phosphoglycerate, PEP phosphoenolpyruvate, PYR pyruvate, R5UP ribulose 5-phosphate, X5P xylulose-5-phosphate, R5P ribose-5-phosphate, S7P sedoheptulose-7-phosphate, E4P erythrose-4-phosphate, DAHP 3-deoxy-d-arabinoheptulosonate 7-phosphate, EPSP 5-enolpyruvylshikimate-3-phosphate, Trp tryptophan, PPY phenylpyruvate, HPP 4-hydroxyphenylpyruvate, PAC phenylacetaldehyde, pPAC p-hydroxyphenylacetaldehyde, Phe phenylalanine, Tyr tyrosine, PAA phenylacetic acid, pPAA p-hydroxyphenylacetic acid, PET phenylethanol, pPET p-hydroxyphenylethanol; b Heatmap of the tyrosine biosynthetic related genes in NK14. The map indicates the gene expression levels as the log2 of their fpkm. A decrease in the expression is indicated by the intensity of blue color and an increase by the intensity of red color. The intensity of the color represents the fold value as indicated by the scale shown to the right-hand side. The results are the averages of three biological duplicates
ARO8 and ARO9 catalyze the transfer of an amine group to PPY or HPP to yield l-phe or l-tyr, while ARO10 and PDC5 catalyze the competitive pathways, leading to the production of phenylacetaldehyde and 4-hydroxyphenylacetaldehyde. Elimination of ARO10, PDC5, and PDC6 decreases loss of tyrosine to the Ehrlich pathway (Koopman et al. 2012; Gold et al. 2015). ARO8 was upregulated, while ARO10 and PDC6 were downregulated, consistent with expectations. ARO80 can activate transcription of the genes ARO9 and ARO10 to initiate catabolism of aromatic amino acids in the presence of a poor nitrogen source (Suastegui et al. 2017). Downregulation of ARO80 in NK14 also reduces loss of tyrosine. The deletion of ARO80 did result in a slight improvement in p-CA production (Supplementary Fig. 2). However, the ARO9 gene was slightly downregulated and PDC5 was upregulated, which could represent an option for further modification (Gold et al. 2015).
Gold et al. (2015) suggested that deletion of mitochondrial malate dehydrogenase (MDH1, MDH2, and MDH3) and knockout of pyruvate carboxylase (PYC1) could completely prevent the production of oxaloacetate during growth on glucose. Downregulation of MDH1, MDH2, MDH3, and PYC1 in NK14 confirmed this theory and these genes could be new targets in modification of the tyrosine pathway.
Furthermore, a previous study showed that knockout of some amino acid transporters triggered higher p-CA production (Rodriguez et al. 2017). Genomic region deletion resulted in changes in the transcription levels of some amino acid transporters (Table 2). DEGs related to amino acid metabolism in NK14 are listed in Table 2.
Discussion
Deletion of single genes within the LGR deleted in NK14 did not improve p-CA yield. The creation of desired mutants through LGR deletion provides a new insight into the enhancement of metabolite production. Transcriptome analysis of NK14 provided few explanations for the higher p-CA yield.
Condensation of PEP and E4P is the first step in the tyrosine biosynthetic pathway, so PEP and E4P are important precursors. However, the carbon flux to E4P is much lower than that to PEP under aerobic conditions (Vaseghi et al. 1999). In NK14, RKI1 and TKL1 were upregulated, while ZWF1 was downregulated; this could serve as a balance between the two substrates (Deaner and Alper 2017). Almost all the genes involved in the glycolysis pathway were upregulated after LGR deletion; this finding suggests an additional direction for modification of energy metabolism other than the amino acid biosynthetic pathway.
Deletion of the transcription factor ARO80 led to a slight improvement in p-CA production compared to BY4741. One explanation is that ARO9 and ARO10 are also under the control of nitrogen catabolite repression and are highly induced by aromatic amino acids besides transcription factor ARO80 (Godard et al. 2007; Lee and Hahn 2013). The lower gene expression levels for the amino acid transporters (AVT6, VBA5, and THI72) also provide a new idea for tyrosine pathway modification other than overexpression of rate-limiting enzymes and elimination of a competitive branch or feedback inhibition. Identification of a potential transcriptional regulator could significantly help in rewiring of carbon distribution to increase carbon entry into the tyrosine pathway.
In conclusion, NK14 is a mutant with enhanced tyrosine production and could serve as a platform strain for further genetic modification. The transcriptome analysis verified our results and points to new genetic modification candidates.
References
Deaner M, Alper HS (2017) Systematic testing of enzyme perturbation sensitivities via graded dCas9 modulation in Saccharomyces cerevisiae. Metab Eng 40:14–22
Eichenberger M, Lehka BJ, Folly C, Fischer D, Martens S, Simon E, Naesby M (2017) Metabolic engineering of Saccharomyces cerevisiae for de novo production of dihydrochalcones with known antioxidant, antidiabetic, and sweet tasting properties. Metab Eng 39:80–89
Galanie S, Thodey K, Trenchard I, Interrante MF, Smolke C (2015) Complete biosynthesis of opioids in yeast. Science 349:1095–1100
Giaever G, Chu AM, Ni L et al (2002) Functional profiling of the Saccharomyces cerevisiae genome. Nature 418:387–391
Godard P, Urrestarazu A, Vissers S, Kontos K, Bontempi G, van Helden J, Andre B (2007) Effect of 21 different nitrogen sources on global gene expression in the yeast Saccharomyces cerevisiae. Mol Cell Biol 27:3065–3086
Gold ND, Gowen CM, Lussier FX, Cautha SC, Mahadevan R, Martin VJ (2015) Metabolic engineering of a tyrosine-overproducing yeast platform using targeted metabolomics. Microb Cell Fact 14:73
Hong KK, Nielsen J (2012) Metabolic engineering of Saccharomyces cerevisiae: a key cell factory platform for future biorefineries. Cell Mol Life Sci 69:2671–2690
Koopman F, Beekwilder J, Crimi B et al (2012) De novo production of the flavonoid naringenin in engineered Saccharomyces cerevisiae. Microb Cell Fact 11:155
Lee K, Hahn JS (2013) Interplay of Aro80 and GATA activators in regulation of genes for catabolism of aromatic amino acids in Saccharomyces cerevisiae. Mol Microbiol 88:1120–1134
Lehka BJ, Eichenberger M, Bjorn-Yoshimoto WE et al (2017) Improving heterologous production of phenylpropanoids in Saccharomyces cerevisiae by tackling an unwanted side reaction of Tsc13, an endogenous double-bond reductase. FEMS Yeast Res 17:1–12
Li M, Kildegaard KR, Chen Y, Rodriguez A, Borodina I, Nielsen J (2015) De novo production of resveratrol from glucose or ethanol by engineered Saccharomyces cerevisiae. Metab Eng 32:1–11
Liu Q, Wu Y, Yang P, Zhang X, Bai Y, Xu H, Qiao M (2014) mazF-mediated deletion system for large-scale genome engineering in Saccharomyces cerevisiae. Res Microbiol 165:836–840
Luttik MA, Vuralhan Z, Suir E, Braus GH, Pronk JT, Daran JM (2008) Alleviation of feedback inhibition in Saccharomyces cerevisiae aromatic amino acid biosynthesis: quantification of metabolic impact. Metab Eng 10:141–153
Mao J, Liu Q, Song X, Wang H, Feng H, Xu H, Qiao M (2017) Combinatorial analysis of enzymatic bottlenecks of l-tyrosine pathway by p-coumaric acid production in Saccharomyces cerevisiae. Biotechnol Lett 39:977–982
Rodriguez A, Chen Y, Khoomrung S, Ozdemir E, Borodina I, Nielsen J (2017) Comparison of the metabolic response to over-production of p-coumaric acid in two yeast strains. Metab Eng 44:265–272
Suastegui M, Yu Ng C, Chowdhury A et al (2017) Multilevel engineering of the upstream module of aromatic amino acid biosynthesis in Saccharomyces cerevisiae for high production of polymer and drug precursors. Metab Eng 42:134–144
Vaseghi S, Baumeister A, Rizzi M, Reuss M (1999) In vivo dynamics of the pentose phosphate pathway in Saccharomyces cerevisiae. Metab Eng 1:128–140
Acknowledgements
This work was supported by Tianjin Research Program of Application Foundation and Advanced Technology of China (CN) (17JCZDJC32200).
Supplementary Information
Supplementary Table 1—The plasmids used in this study
Supplementary Table 2—Strains with single gene knock-out used in this study (Giaever et al. 2002)
Supplementary Table 3—The primers used in this study
Supplementary Fig. 1—p-CA synthesis of single gene deletion strains. The tyrosine yield was measured through the p-coumaric acid by strains with the TAL plasmid. The strains were cultivated at 30 °C in 50 mL SC-his-tyr medium for 72 h fermentation. Error bars represent the standard deviation of three biological duplicates
Supplementary Fig. 2—p-CA synthesis of ARO80 deletion strain. Tyrosine yield was measured every 24 h during the fermentation through the p-coumaric acid by strains with the TAL plasmid. The strains were cultivated at 30 °C in 15 mL SC-his-tyr medium in flasks. Error bars represent the standard deviation of three biological duplicates
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Wu, Y., Cai, M., Song, X. et al. Comparative transcriptome analysis of genomic region deletion strain with enhanced l-tyrosine production in Saccharomyces cerevisiae. Biotechnol Lett 42, 453–460 (2020). https://doi.org/10.1007/s10529-019-02784-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10529-019-02784-1