
Abstract
Objectives
To improve the stability and sweetness of the sweet-tasting protein, monellin, by using site-directed mutagenesis and a Pichia pastoris expression system with a GAPDH constitutive promoter.
Results
Both wild-type and E2 N mutant of single-chain monellin gene were cloned into the PGAPZαA vector and expressed in Pichia pastoris. The majority of the secreted recombinant protein, at 0.15 g/l supernatant, was monellin. This was purified by Sephadex G50 chromatography. The sweetness threshold of wild-type and E2 N were 30 μg/ml and 20 μg/ml, respectively. Compared with the proteins expressed in Escherichia coli, the thermostability of both proteins was improved. The N-terminal sequence is determinative for the sweetness of the proteins expressed in yeast strains.
Conclusions
Site-directed mutagenesis, modification of the N-terminus of monellin, and without the need of methanol induction in P. pastoris expression system, indicate the possibility for large-scale production of this sweet-tasting protein.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Monellin, a sweet protein extracted from the West African plant, Dioscoreophyllum cumminsii, has a high sweetness and low calorific value (Morris et al. 1973). It consists of 94 amino acids in two chains (A and B) that are held together by noncovalent interactions. However, monellin loses its sweet activity at 50 °C. To improve its stability, single-chain monellin (MNEI) protein was created in which the two natural chains are joined via a Gly-Phe dipeptide linker and this retained the same activity and sweetness as the native protein at extreme pH and high temperatures (Kim et al. 1989). Compared with other chemical sweeteners, monellin has no toxic side effects and can be regarded as a sweet additive for cardiovascular, diabetic and hyperlipidemia patients who are related to the consumption of sugars. So far Japan is the only country that has approved it as a food additive (Faus 2000).
Monellin has been expressed in Escherichia coli, Saccharomyces cerevisiae, Bacillus subtilis, the food yeast Candida utilis and some plants (Chen et al. 2005, 2007, 2011; Kondo et al. 1997; Lee et al. 2012; Liu et al. 2015). However, its propensity to denaturate at high temperatures has limited its applications (Faus 2000). To overcome this, mutagenesis studies have been performed to improve its sweetness and stability. For example, the Y65R mutant exhibits superior sweetness and significant thermal stability, suggesting its potential as an additive in beverages (Rega et al. 2015). A stable variant E23A of MNEI has been over-expressed in tobacco chloroplasts and shows retained sweetness and enhanced stability above 70 °C (Lee et al. 2012). We have expressed and purified two variants, C41A and E23A, in E. coli with increased sweetness and stability, respectively (Liu et al. 2016). Nevertheless, no variant of the sweet-tasting protein has been expressed in food yeast strains until now.
Pichia pastoris is an efficient, productive and economic eukaryotic expression host for both secretion and intracellular expression on an industrial scale (Weinacker et al. 2014). The FDA (Food and Drug Administration) has approved it as a GRAS (generally recognized as safe) expression system. Furthermore, the GAPDH (glyceraldehyde-3-phosphate dehydrogenase) promoter (P GAP ) is a constitutive type promoter isolated from P. pastoris. Expression of target protein under control of P GAP in glucose-grown cells was higher than that under control of the commonly employed alcohol oxidase 1 promoter (P AOX1 ) in methanol-grown cells (Waterham et al. 1997). Moreover, there is no need to add any inducing agent in the process of protein production. Therefore, the fermentation process under control of P GAP is easier than that of P AOX1 . To promote the large-scale production of monellin, we expressed and purified both wild-type and E2 N mutant of MNEI in P. pastoris and evaluated their yield, sweetness and stability.
Materials and methods
Strains, plasmids and chemicals
E. coli DH5α, Pichia pastoris GS115 and plasmid PGAPZαA were used. (The latter was from Invitrogen.) EasyPfu DNA Polymerase, restriction enzymes XhoI, NotI and AvrII and T4 DNA ligase were from Beijing TransGen Biotech Co. Molecular manipulation kits were from Takara Bio (Dalian, China). All chemicals were analytical grade and obtained from Sangon Biotech (Shanghai, China).
Culture medium
E. coli was cultured on the low salt LB (5 g/l yeast extract, 10 g/l tryptone, 5 g/l NaCl, pH 7) medium containing 25 μg zeocin/ml. P. pastoris strains were cultured and screened on the YPD medium(5 g/l yeast extract, 10 g/l tryptone, 10 g/l glucose, pH 6)containing 100 μg zeocin/ml.
Construct
To facilitate expression in P. pastoris, the full coding sequence of the MNEI was optimized based on the Codon Adaptation Tool (http://www.jcat.de/), and the gene was synthetized by Taihe Biotechnology Co., Ltd. The gene and the plasmid PGAPZαA were digested with XhoI and NotI and then ligated with the T4 DNA ligase. The ligated product was transformed into the host E. coli DH5α to select the correct recombinants. The recombinant plasmid was designated as PGAPZαA-MNEI and verified by DNA sequencing.
Site-directed mutagenesis
The mutated E2 N gene was amplified by PCR using a forward primer (5′-CCCTCGAGAAAAGAGAGGCTGAAGCTGGTAACTGGGAG-3′) and a reverse primer (5′-TTGCGGCCGCTTAGGGAGGAGGCACAGGTCCGTTG-3′) (the underlined sequences indicates XhoI and NotI sites, and the bold codon indicates the mutated residue site). The PCR product was purified with a PCR purification kit and digested with XhoI and NotI, and then cloned into the plasmid PGAPZαA. The plasmid harboring mutated gene was verified by DNA sequencing.
Transformation, expression and purification of the wild-type and mutated MNEI
The preparation of competent P. pastoris cells was as described previously (Poirier et al. 2012). The gene construct (5–20 μg) was linearized with AvrII and mixed with 80 μl P. pastoris competent cells, and then were treated with electric shock at 1.5 kV,25 μF and 200 Ω. The transformants were selected on the YPD plate supplemented with 182 g sorbitol/l and 100 μg zeocin. The genome of P. pastoris was extracted, followed by PCR to select the positive transformants. To over-express the protein, the recombinant P. pastoris strains were inoculated into 200 ml YPD broth, and then shaken at 250 rpm and 30 °C for 3 days. The culture was centrifuged at 10,000×g for 15 min, and the supernatant was concentrated by salting out method. After desalting by dialysis in MilliQ water, the recombinant protein was purified by Sephadex G50 column equilibrated with 50 mM citrate buffer (pH 6). Recombinant MNEI was eluted as a single peak. Protein concentration was determined by Bradford method. The MNEI expressed in E. coli was purified as described previously (Liu et al. 2016).
Sweet threshold assay
Double-blind taste assays were performed by a panel of ten healthy volunteer tasters, five males and five females, 20–60 years old (Liu et al. 2016).
Thermostability
To investigate the heat stability, the proteins expressed in P. pastoris and E. coli were concentrated by ultrafiltration at the same final concentrations (100 μg/ml). Subsequently, 100 μl protein solution was held at 60, 65, 70, 75 and 80 °C for up to 8 h. The heated proteins were taken at various times (2 h once), and then centrifuged to remove the sediment. Supernatant solutions were analyzed by SDS-PAGE to evaluate denaturation or aggregation. The sweetness of the preheated proteins were evaluated as described above. Each experiment were carried out in triplets and the results were averaged.
Results
Construction of the wild-type and mutant expression-secretion vectors
To facilitate the expression in P. pastoris, the codons of MNEI was optimized and a 294 bp nucleotide sequence was obtained (Fig. 1), in which the start condon ATG coding Met was removed. Therefore, the resulted protein had an initial N-terminal Gly after cleavage of the α-factor signal peptide. For the E2 N variant, the codon CCG coding Glu was changed to AAC coding Asn. The expression plasmids PGAPZαA harboring the wild-type or mutated E2 N gene under the control of a strong constitutive GAPDH promoter were constructed, which were then transformed into P. pastoris, and the correct transformants were verified by PCR (Fig. 2).

Nucleotide and amino acid sequence of single-chain monellin (MNEI) construct. The mutated E2 N site was boxed

PCR amplification of the 4 positive transformants in Pichia pastoris. Two universal primers of PGAPZαA vector (forward: pGAP primer; reverse: 3′-AOX1 primer) were used in PCR with the P. pastoris genome as the template. 50 μl PCR reaction was subjected to 30 cycles (30 s at 94 °C; 30 s at 45 °C; 1 min at 72 °C). Lane M: DNA markers. Lanes 1–4: the amplified PCR products
Expression and purification of the recombinant MNEI
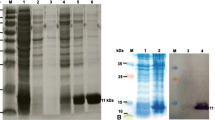
The over-expressed protein was secreted into the culture medium by the α-factor signal peptide. There were few other host proteins in the culture supernatant (Fig. 3). The proteins were further purified by Sephadex G50 chromatography. Notably, the P. pastoris expressed protein showed a slight lower gel band than the E. coli expressed protein, presumably due to absence of the N-terminal His-tag and the initial Met (Fig. 3). The protein produced in P. pastoris reached 0.15 g/l in this shake-flask cultivation.

SDS-PAGE analysis of the expression of MNEI in E. coli, and MNEI and E2 N variant in P. pastoris. Lane M: molecular weight marker. Lane 1: total soluble protein in P. pastoris of MNEI. Lane 2: supernatant in P. pastoris of MNEI. Lane 3: the purified MNEI. Lane 4: total soluble protein in P. pastoris of E2 N. Lane 5: supernatant in P. pastoris of E2 N. Lane 6: the purified E2 N. Lane 7: total soluble protein in E. coli of MNEI. Lane 8: supernatant in E. coli of MNEI. Lane 9: the purified MNEI in E. coli
Sweet threshold assay
Table 1 shows that the sweetness of mutant E2 N was improved to be approx. 1.5 times sweeter than that of the wild-type. Both of the proteins displayed strong sweetness which were about 300–500 times sweeter than sucrose on a weight basis.
Thermostability
Figure 4a shows that MNEI, expressed in E. coli, denatured after heating at 65 °C for 6 h. However, both wild-type and E2 N expressed in P. pastoris retained solubility under the same conditions (Fig. 4b, lanes 1 and 2). These results indicate that the proteins expressed in P. pastoris had better stability than those expressed in E. coli.

Heat stability of MNEI and E2 N variant. a SDS-PAGE analysis of the thermostability of MNEI expressed in E. coli. Lane M: molecular weight marker. Lane 1: MNEI without heat treatment. Lane 2: MNEI heat treatment at 65 °C for 2 h. Lane 3: MNEI heat treatment at 65 °C for 4 h. Lane 4: MNEI heat treatment at 65 °C for 6 h. Lane 5: MNEI heat treatment at 70 °C for 6 h. Lane 6: MNEI heat treatment at 75 °C for 6 h. Lane 7: MNEI heat treatment at 80 °C for 8 h. b SDS-PAGE analysis of the thermostability of MNEI and E2 N expressed in P. pastoris. Lane M: molecular weight marker. Lane 1: MNEI heat treatment at 65 °C for 6 h. Lane 2: E2 N heat treatment at 65 °C for 6 h. Lane 3: MNEI heat treatment at 70 °C for 6 h. Lane 4: E2 N heat treatment at 70 °C for 6 h. Lane 5: MNEI heat treatment at 75 °C for 6 h. Lane 6: E2 N heat treatment at 75 °C for 6 h. Lane 7: MNEI heat treatment at 80 °C for 8 h. Lane 8: E2 N heat treatment at 80 °C for 8 h
Discussion
The single-chain monellin protein was expressed in P. pastoris, which will be a substantial advantage for high density fermentation and large scale industrial production. Monellin has been heterologously expressed in P. pastoris and other hosts. However, the need of methanol induction increases the probability of risk. In our expression system, the P GAP (glyceraldehyde-3-phosphate dehydrogenase promoter) is a constitutive type promoter (Waterham et al. 1997), thus there is no need to add any inducing agent in the process of protein production. Furthermore, the codon of monellin gene was optimized according to the bias of P. pastoris, and the expression-secretion vector PGAPZαA harboring the α-factor signal peptide of Saccharomyces cerevisiae was constructed, which can secrete the recombinant protein into the culture medium. The majority of the extracellular proteins from P. pastoris was monellin, which makes the purification much easier than other expression systems reported previously (Chen et al. 2011). These advantages can facilitate the large scale industrial production of this protein.
Previous studies have not evaluated the sweetness threshold of monellin when expressed in yeast strains or the recombinant proteins were tasteless (Chen et al. 2011; Kondo et al. 1997; Liu et al. 2015). Our study showed that removal of the N-terminal Met of recombinant MNEI made it reassemble as the native protein; the exact evaluation of its sweetness threshold was thus possible, suggesting the critical role of N-terminus for the sweetness of recombinant monellin protein in yeast strains. However, the proteins expressed in P. pastori are not as sweet as those expressed in E. coli. There may exist a small change in structure of the protein produced in eukaryotic expression system compared with those in prokaryote due to glycosylation modification (De Pourcq et al. 2010), which could affect the sweetness. On the other hand, the sweetness of E2 N is improved than that of wild-type. Glu 2 is located at the N-terminal surface of the protein (Supplementary Fig. 1), and replacement of this negative residue by the neutral Asn decreases the negative charge of protein surface, which is involved in the interaction with the sweet taste receptor, thereby increasing the sweetness (Liu et al. 2012). Furthermore, the intramolecular hydrogen or hydrophobic interactions network could be rearranged due to the E2 N mutant and might lead to a change of conformation or folding of the protein, thus affecting the sweetness (Templeton et al. 2011).
The slow commercial development of monellin is mainly due to its instability at high temperatures. The protein expressed in P. pastoris displays improved stability than those expressed in E. coli. Glycosylation modification in eukaryotic organisms can improve the stability of cell wall and some proteins in yeast through N-glycosylation and O-mannosylation (De Pourcq et al. 2010; Willer et al. 2005). Moreover, a correlation between the thermostability and the function of the protein was found. Further research is needed to optimize the production conditions of large-scale industrialization. Taken together, our study demonstrates that the properties of the sweet-tasting protein monellin can be improved by gene mutagenesis and sequence optimization in a non-induced P. pastoris expression system.
References
Chen Z, Cai H, Lu F, Du L (2005) High-level expression of a synthetic gene encoding a sweet protein, monellin, in Escherichia coli. Biotechnol Lett 27:1745–1749
Chen Z, Heng C, Li Z, Liang X, Xinchen S (2007) Expression and secretion of a single-chain sweet protein monellin in Bacillus subtilis by sacB promoter and signal peptide. Appl Microbiol Biotechnol 73:1377–1381
Chen Z, Li Z, Yu N, Yan L (2011) Expression and secretion of a single-chain sweet protein, monellin, in Saccharomyces cerevisiae by an α-factor signal peptide. Biotechnol Lett 33:721–725
De Pourcq K, De Schutter K, Callewaert N (2010) Engineering of glycosylation in yeast and other fungi: current state and perspectives. Appl Microbiol Biotechnol 87:1617–1631
Faus I (2000) Recent developments in the characterization and biotechnological production of sweet-tasting proteins. Appl Microbiol Biotechnol 53:145–151
Kim SH, Kang CH, Kim R, Cho JM, Lee YB, Lee TK (1989) Redesigning a sweet protein: increased stability and renaturability. Protein Eng 2:571–575
Kondo K, Miura Y, Sone H, Kobayashi K, Iijima H (1997) High-level expression of a sweet protein, monellin, in the food yeast Candida utilis. Nat Biotechnol 15:453–457
Lee S-B, Kim Y, Lee J, Oh K-J, Byun M-O, Jeong M-J, Bae S-C (2012) Stable expression of the sweet protein monellin variant MNEI in tobacco chloroplasts. Plant Biotechnol Rep 6:285–295
Liu B, Ha M, Meng XY, Khaleduzzaman M, Zhang Z, Li X, Cui M (2012) Functional characterization of the heterodimeric sweet taste receptor T1R2 and T1R3 from a New World monkey species (squirrel monkey) and its response to sweet-tasting proteins. Biochem Biophys Res Commun 427:431–437
Liu J, Yan DZ, Zhao SJ (2015) Expression of monellin in a food-grade delivery system in Saccharomyces cerevisiae. J Sci Food Agric 95:2646–2651
Liu Q, Li L, Yang L, Liu T, Cai C, Liu B (2016) Modification of the sweetness and stability of sweet-tasting protein monellin by gene mutation and protein engineering. Biomed Res Int 2016:3647173
Morris JA, Martenson Deibler RG, Cagan RH (1973) Characterization of monellin, a protein that tastes sweet. J Biol Chem 248:534–539
Poirier N, Roudnitzky N, Brockhoff A, Belloir C, Maison M, Thomas-Danguin T, Meyerhof W, Briand L (2012) Efficient production and characterization of the sweet-tasting brazzein secreted by the yeast Pichia pastoris. J Agric Food Chem 60:9807–9814
Rega MF, Di Monaco R, Leone S, Donnarumma F, Spadaccini R, Cavella S, Picone D (2015) Design of sweet protein based sweeteners: hints from structure–function relationships. Food Chem 173:1179–1186
Templeton CM, Ostovar pour S, Hobbs JR, Blanch EW, Munger SD, Conn GL (2011) Reduced sweetness of a monellin (MNEI) mutant results from increased protein flexibility and disruption of a distant poly-(l-proline) II Helix. Chem Senses 36:425–434
Waterham HR, Digan ME, Koutz PJ, Lair SV, Cregg JM (1997) Isolation of the Pichia pastoris glyceraldehyde-3-phosphate dehydrogenase gene and regulation and use of its promoter. Gene 186:37–44
Weinacker D, Rabert C, Zepeda AB, Figueroa CA, Pessoa A, Farías JG (2014) Applications of recombinant Pichia pastoris in the healthcare industry. Braz J Microbiol 44:1043–1048
Willer T, Brandl M, Sipiczki M, Strahl S (2005) Protein O-mannosylation is crucial for cell wall integrity, septation and viability in fission yeast. Mol Microbiol 57:156–170
Acknowledgments
This work was supported by the Natural Science Foundation of China (31271118 and 31501450).
Supporting Information
Supplemental Fig. 1—The three-dimensional structure of monellin (PDB:1IV9). The α-helix, β-sheet and β-loop are colored in red, yellow and green, respectively. The mutated residue Glu 2 at the protein surface is labeled, rendered as sticks and colored by atomic types.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Cai, C., Li, L., Lu, N. et al. Expression of a high sweetness and heat-resistant mutant of sweet-tasting protein, monellin, in Pichia pastoris with a constitutive GAPDH promoter and modified N-terminus. Biotechnol Lett 38, 1941–1946 (2016). https://doi.org/10.1007/s10529-016-2182-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10529-016-2182-4