
Abstract
Objective
To compare stably-transfected Drosophila melanogaster S2 and mammalian Chinese hamster ovary (CHO) cells for the expression and secretion efficiency of biologically-active human coagulation factor IX (hFIX).
Result
Selection of an hFIX-expressing cell line derived from stably-transfected S2 cells was performed over 2 weeks, while the same procedure required 2 months for stably-transfected CHO cells. Furthermore, the selected S2 cell line was superior in producing of total hFIX protein (70 % increase), biologically-active hFIX (35 % increase), and specific hFIX activity (20 % increase) relative to the selected CHO cell line. Enrichment for functional, fully γ-carboxylated hFIX species via barium citrate adsorption demonstrated that up to 90 % of the hFIX expressed by S2 cells was γ-carboxylated versus 79 % of CHO-expressed hFIX. Inhibition of N-glycosylation by tunicamycin indicated that N-glycosylation of S2-expressed hFIX had occurred to a similar extent as in the CHO-produced hFIX.
Conclusion
The Drosophila S2 cell system is an attractive candidate for the efficient production of recombinant hFIX as it has the potential of significantly reducing the cell development time, while producing functional hFIX.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Blood coagulation human factor IX (hFIX) is a vitamin K-dependent (VKD) glycoprotein that is synthesized by the liver and circulates in blood as an inactive zymogen. Once activated, FIXa associates with its cofactor, VIIIa, on an anionic membrane surface and subsequently catalyzes the activation of blood coagulation factor X, an important step in the intrinsic pathway of the blood coagulation cascade (Bos et al. 2016). A deficiency or functional defect in FIX results in hemophilia B, which is an X-linked bleeding disorder that occurs in 1/30,000 males. The standard treatment regimen for hemophilia B consists of protein replacement therapy with either plasma-derived or recombinant FIX. A drawback of these approaches is that plasma-derived material poses an inherent risk of transmission with blood-borne diseases, while recombinant protein production requires an expression system that allows for the efficient generation of high quantities of functional protein.
The majority of all biopharmaceuticals, including recombinant FIX, are currently produced by mammalian cell lines since these are able to generate proteins with biochemical characteristics similar to those naturally occurring in humans. A distinct disadvantage of the mammalian cell-based expression systems is the fact that extensive and time-consuming screening is required to identify high producing clones (Nair et al. 2011). Moreover, once generated, the stable producer line needs to be maintained under selective pressure and, even under these conditions, protein expression may be unstable over prolonged culture (Vatandoost et al. 2012).
A cell system that has been used increasingly for the expression of various recombinant proteins is that based on Drosophila melanogaster Schneider line 2 (S2) cells, which offers a straightforward approach to stably produce high quantities of functional protein (Moraes et al. 2012). The S2 expression system has the advantage of proliferating without requiring CO2, being easily adapted to large-scale fermenters, being capable of long-term continuous culture, having a null expression background, and having the appropriate post-translational machinery (Moraes et al. 2012; Vatandoost et al. 2012). Furthermore, Drosophila cells can integrate up to 1000 copies of an expression cassette in a single transfection event, while protein expression remains tightly regulated even at high copy numbers with low basal expression maintained in the absence of an inducer (Kim et al. 2008). Using this system, a wide variety of proteins that are appropriately processed and maintain biological activity have successfully been expressed (Moraes et al. 2012).
We have demonstrated that Drosophila S2 cells are capable of expressing biologically-active human FIX (Vatandoost et al. 2012). To express full functionality, human FIX requires several complex post-translational modifications, including γ-carboxylation of twelve N-terminal glutamic acid residues, N- and O-linked glycosylation, and proteolytic processing of the signal peptide and the propeptide (Bos et al. 2016; Hansson and Stenflo 2005). S2 cells are capable of these types of modifications which are basically similar to those in mammalian cells (Percival et al. 1997). As such, the S2 cell system provides an attractive alternative for mammalian recombinant protein expression. In the current study, we examined the generation of functional human FIX-producing insect Drosophila S2 and mammalian CHO cell lines that stably produce human FIX and compared the clone selection in terms of timing as well as quantitative and qualitative FIX expression.
Materials and methods
Oligonucleotides were synthesized by Bioneer. All cell culture reagents and the Drosophila Schneider (S2) cells were from Thermo Fisher Scientific, except Schneider’s insect medium, penicillin G, and streptomycin (Sigma-Aldrich). The RNA preparation kit (Tripure), reverse transcriptases (RT; M-MuLV), geneticin (G418), and protease inhibitors were purchased from Roche.
Factor IX expression in CHO cells
CHO cells (obtained from the Pasteur Institute, Iran) were cultured and transfected as described previously (Haddad-Mashadrizeh et al. 2009; Vatandoost et al. 2012). Briefly, the CHO cells were grown in 5 % CO2 at 37 °C, subcultured at 2 × 105 cells in 2 ml in 6-well plates, and transfected with 2 µg pcDNA3-hFIX using FuGene–6. Individual clones were expanded in 450 µg geneticin/ml. Expression media containing 6 µg vitamin K1/ml and 10 % (v/v) FBS was added to ~70 % confluent cells, upon which the individual clones were screened for FIX production.
Factor IX expression in S2 cells
Generation of the human FIX expressing plasmid pMT-hFIX has been described (Vatandoost et al. 2012). S2 cells were maintained at 28 °C under normal atmosphere in Schneider’s insect medium supplemented with penicillin (50 units/ml) and streptomycin (50 µg/ml). One day before transfection, 3 × 106 cells were seeded in 3 ml in 6-well plates, upon which the cells were allowed to loosely adhere. S2 cells were transfected with pMT-hFIX and the pCoHygro plasmid that comprises the hygromycin resistance gene employing the calcium phosphate co-precipitation method with minor modifications (Cherbas and Cherbas 2007). After 48 h, the cells were cultured in 300 μg hygromycin B/ml. Cell density and viability was monitored prior to and following transfection by Trypan.
Blue exclusion using a 0.4 % (w/v) solution. To select clones that stably express FIX, two strategies were employed. In the first, single cell strategy, approx. 150 × 106 parental S2 cells were treated for 4 h with mitomycin C to induce cell cycle arrest, upon which these feeder cells were plated in 24-well plates at 3 × 106 cells/ml (Nilsen and Castellino 1999). Subsequently, the FIX-transfected cells were added at 1 cell/well. After approx. 2 weeks, the single cell-derived clones were examined for FIX production. In the second, limiting dilution approach, the transfected cells were transferred to 96-well plates at 5 × 104 cells per 100 µl. 2 weeks after cell transfer, the clones were screened for FIX expression following induction with 0.5 mM CuSO4 in the presence of 6 µg vitamin K1/ml. Mock-transfected cells did not survive 10 days post-transfection.
Quantitative and qualitative analysis of human factor IX
Human FIX was quantified in conditioned media employing an ELISA (Asserachrom IX:Ag, Stago, France) following the procedure provided by the manufacturer. In addition, intracellularly accumulated FIX was assessed, for which the cells were pelleted by centrifugation at 100×g for 5 min, upon which they were resuspended in 500 µl ice-cold lysis buffer (100 mM KCl, 2 mM MgCl2, 10 mM, HEPES pH 7.5, 0.5 % Triton ×100) containing an antiprotease mix (complete Protease Inhibitor, Roche). Subsequently, the lysate was centrifuged at 12,000×g for 10 min. at 4 °C, upon which the supernatant was assessed for FIX. The functional activity of FIX was examined using an aPTT assay (Vatandoost et al. 2012). Briefly, human plasma immuno-depleted of FIX (100 µl; Stago) was mixed with conditioned media (100 µl) and aPTT reagent (100 µl; BioMerieux). After 3 min of incubation at 37 °C, 100 µl prewarmed CaCl2 (25 mM) was added to the mixture, and the clotting time was recorded. Normal human plasma (Iranian Blood Transfusion Organization) was used as reference, and one unit of FIX activity corresponds to the amount of FIX in 1 ml of normal plasma (~5 μg/ml).
RNA preparation and RT-PCR Analysis
Total RNA from 106 cells expressing human FIX was extracted according to the protocol provided by the manufacturer (Tripure kit). First-strand cDNA synthesis was performed using random primers and M-MuLV reverse transcriptase (Sinaclone, Iran). The generated fragments were subsequently used as template for the PCR-amplification of the double stranded cDNA, corresponding to a section of the hFIX coding sequence.
Quantification of γ-carboxylated factor IX
Barium citrate precipitation of γ-carboxylated human FIX was adapted from previously described methods (Yao et al. 1991). Briefly, sodium citrate (14 mg) and 1 M BaCl2 (95 µl) were added to 2 ml of conditioned media, which were incubated for 1 h at 4 °C with gentle mixing. The mixture was centrifuged at 100×g for 5 min, and the supernatant was kept for the analysis of unabsorbed FIX lacking γ-carboxyglutamyl residues. The pelleted γ-carboxylated FIX was washed with ice-cold 5 mM BaC12, subsequently centrifuged at 100×g for 5 min, resuspended in 0.1 M sodium citrate (1 ml), and adjusted to 10 % (v/v) ammonium sulphate to dissolve the adsorbed hFIX. After 30 min at 0 °C and subsequent centrifugation at 100×g for 5 min after which the BaSO4 pellet was discarded, FIX was determined in the supernatant by ELISA as described.
Glycoprotein analysis
To study glycoprotein synthesis, the FIX-expressing S2 and CHO cells were cultured in the absence or presence (5 µg/ml) of tunicamycin. After 24 h, the conditioned media was treated with barium citrate, and the FIX adsorpted was determined by ELISA as described. Subsequently, the adsorpted FIX (3 µg) was analyzed by SDS PAGE using precast 4–12 % gradient gels (Invitrogen) under reducing conditions (50 mM dithiothreitol) in the MES buffer system.
Statistical analysis
The ANOVA program for analysis of variance followed by a Tukey post hoc test was performed for evaluation; P < 0.05 was considered statistically significant. All statistical analyses were carried out with SPSS 16 (SPSS Inc., Chicago, IL, USA).
Results
Selection of clones stably expressing factor IX
The selection of single cell-derived CHO clones that stably express human FIX was performed over a period of 2 months and resulted in the selection of twelve high-producer clones. Comparison of the FIX expression following expansion of these clones in a 25 cm2 flask to 95 % confluency revealed the four clones with highest FIX production, of which clone 8 expressed FIX with the highest specific activity (0.43 U/mg; Table 1).
Because S2 cells do not survive when cultured at lower than 2 × 105 cells/ml, simply diluting them at a theoretical dilution of 1 cell/well was not a feasible approach. Therefore, two strategies were employed to overcome this issue. In the first strategy, FIX-expressing S2 clones were derived from single cells as a result of co-cultures with cell cycle-arrested, non-transfected S2 cells. Out of 24 single cell cultures, two clones (1 and 21) demonstrated FIX expression. In the second approach, FIX-expressing clones were generated following limited cell dilutions (5 × 105 cells/ml) of 5 × 104 cells. Out of the 96 cultures that were initiated, 36 clones expressed FIX, from which the twelve with the highest FIX production were expanded into a 48-well plate. Screening for FIX production revealed two high producing clones (clones 2 and 12). Comparison of the FIX expression following 24 h expansion of the four high-producer clones at a seeding density of 6 × 106 cells/ml revealed that clone 1 expressed FIX with the highest specific activity (0.74 U/mg; Table 1).
Overall, these findings demonstrate that FIX-expressing S2 clones were generated in the relative short time span of 2 weeks, while the selection of CHO clones required 2 months. Furthermore, selected S2 cells are not only capable of expressing up to 30 % more human FIX protein as compared to CHO cells (144 vs. 440 ng/ml, Table 1), but the FIX produced has higher functionality, as indicated by the almost 20 % increase in specific activity (0.74 vs. 0.43 U/mg; Table 1). As such, the specific FIX activity exceeds that of FIX circulating in human plasma, which has a specific activity of 0.2 U/mg (Thompson 1986).
Factor IX expression in stably-transfected S2 and CHO cell lines
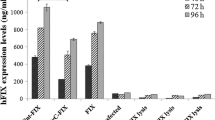
Factor IX (FIX) expression by the CHO and S2 cell lines derived from clones 8 and 1, respectively, was further confirmed employing RT-PCR, which demonstrated the presence of FIX mRNA in both cell types (data not shown). Next, we examined conditioned media and cell lysates for FIX antigen levels. While the CHO cell line expressed up to 62 ng/ml/106 cells FIX after 72 h, FIX expression by the S2 cell line increased with time up to 70 % more and accumulated to 425 ng/ml per/106 cells (Fig. 1a). For both cell lines, intracellular FIX was <12 ng/ml/106 cells. Consistent with these findings, the S2 cell line expressed an up to 35 % higher FIX activities as compared to the CHO cells (63 vs. 18 mU/ml.106 cells). Overall, the S2-expressed FIX displayed a 20 % enhanced specific activity relative to that expressed by the CHO cell line (0.6 vs. 1.1 U/mg; Fig. 1b). For both cell types, the production of functional FIX decreased over 72 h with optimal production at 24 h (Fig. 1). Taken together, these findings show that, in agreement with our observations made during clone selection, the selected S2 cell line produces higher levels of functional human FIX as compared to the CHO cell line.

Factor IX expression in stably-transfected S2 and CHO cell lines. The CHO cell line derived from clone 8 (grey bars) was grown to ~70 % confluency, upon which the media was replaced by expression media comprising 6 µg/ml vitamin K1 and 10 % (v/v) FBS. In the S2 cell line derived from clone 1 (closed bars), FIX expression was induced with 0.5 mM CuSO4 in the presence of 6 µg/ml vitamin K1. At the indicated time points, FIX expression was assessed in the conditioned media by assaying FIX antigen (panel a) or activity levels to determine the specific FIX activity (panel b). The data are the mean ± S.D. of 3 similar experiments
Post-translational modifications of factor IX
The general characteristic of vitamin K-dependent proteins to adsorb to barium citrate via their γ-carboxylated glutamic residues (Jallat et al. 1990) was used to check whether the recombinant cells were capable of proper γ-carboxylation of human FIX. Here, we used barium citrate adsorption to fractionate active γ-carboxylated protein from inactive subpopulations. During this process, neither poorly γ-carboxylated molecules nor non-γ-carboxylated are trapped by barium and remain in the soluble fraction; the barium-bound hFIX can be subsequently eluted from the precipitate (Jallat et al. 1990). Following barium citrate precipitation, the supernatant obtained from the S2 and CHO cell lines was enriched for FIX (Fig. 2). This was confirmed by Western blot analysis and probing with a human FIX-specific antibody, which resulted in detection of the protein band migrating at ~55 kDa only (data not shown). Furthermore, this also demonstrated that some other non-FIX proteins are also enriched through barium citrate adsorption (Fig. 2).

SDS-PAGE analysis of γ-carboxylated factor IX following barium adsorption. Conditioned media from the selected CHO or S2 cell line was collected, and γ-carboxylated FIX was isolated by barium citrate precipitation as described in “Materials and Methods” section. Factor IX (3 μg/lane) was subjected to SDS-PAGE under reducing conditions and visualized by staining with Coomassie Brilliant Blue R-250. Lane 1 represents plasma-derived FIX; lanes 2 and 3, FIX expressed by S2 and CHO cells following barium adsorption (‘+Ba’); lanes 4 and 5, FIX expressed by S2 and CHO cells prior to barium adsorption (‘+Ba’). The apparent molecular weights of the standards are indicated on the left
Assessment of FIX recovery following barium citrate adsorption further corroborated our previous findings and demonstrated that while the overall FIX production increases over time, the portion of functional FIX decreases (Table 2). Up to 90 % of the S2 cell-produced and up to 79 % of the CHO cell-produced FIX were recovered employing barium citrate adsorption, indicating that the majority of the FIX produced was γ-carboxylated.
To study the potential variations in N-glycans on FIX produced by CHO cells versus S2 cells, FIX was expressed both in the absence and presence of tunicamycin (5 µg/ml), which blocks N-glycosylation. SDS-PAGE analysis of FIX isolated from conditioned media employing barium citrate adsorption revealed a similar migration pattern for both cell types, both prior to and following tunicamycin treatment of the cells (Fig. 3). While normal FIX migrates at ~55 kDa, once expressed in the presence of tunicamycin it migrated at approx. 50 kDa. This would correspond with the absence of the two FIX N-glycans of ~2.5 kDa each at residues Asn157 and Asn167 (Hansson and Stenflo 2005).

SDS-PAGE analysis of tunicamycin-treated factor IX. The selected CHO or S2 cell line was cultured in the absence (‘−Tu’) or presence (‘+Tu’) of 5 µg/ml of tunicamycin during 24 h, upon which the conditioned media was treated with barium citrate as described in “Materials and Methods” section. The adsorpted FIX (3 μg/lane) was subjected to SDS-PAGE under reducing conditions and visualized by staining with Coomassie Brilliant Blue R-250. The apparent molecular weights of the standards are indicated on the left
Discussion
The production of recombinant human FIX has been undertaken in a variety of mammalian cell lines; however, the FIX expression levels and long periods of clonal selection and gene amplification are still concerns when moving towards large-scale FIX production for therapeutic purposes. The latter is underscored by the current study in which we generated a clonal, FIX-expressing CHO cell line, which required up to 2 months of selection. In contrast, using the Drosophila melanogaster S2 cell system we were able to generate a clonal cell line in the short time period of 2 weeks. This timescale corroborates with that of previously established Drosophila S2 cell lines expressing erythropoietin or human plasminogen (Lee et al. 2000; Nilsen and Castellino 1999). However, it is important to note that since S2 cells need to be maintained at relatively high cell densities, the strategy employed here to establish a Drosophila S2 cell line is fairly more complex relative to that of the CHO cells. High FIX expression levels are another requirement for successful therapeutic protein production. In our expression systems, the S2 cell line proved to be superior in the production of total FIX protein (70 % increased), functional FIX (35 % higher), and specific FIX activity (20 % enhanced) relative to the selected CHO cell line. The rapid selection of stably-transfected and high producer S2 clones likely stems from the facts that the Drosophila S2 cells are capable of integrating up to approx. 1000 vector copies per cell, have a strong Mtn promoter, and have high maximum specific cell growth rates and cell concentrations (Jorge et al. 2008; Moraes et al. 2012; Vatandoost et al. 2012).
The functional FIX activity levels observed in this study were approx. 0.05 U/ml for the CHO cell line, and 0.15 U FIX activity/ml was expressed by the selected S2 cell line. FIX activity levels reported by others for stably-transfected CHO cells vary around 0.25 U/ml (Kim et al. 2009; Liu et al. 2014). However, in both of these studies FIX was expressed in suspension cultures using spinner bottles or shake-flasks, which dramatically improves the volumetric productivity. Furthermore, in both cases the FIX was co-expressed with PACE/furin enzymes, which effectively remove the FIX pro-peptide during post-translational processing. This may lead to enhanced specific FIX activity as well as more efficient protein secretion. Interestingly, our findings suggest that the FIX produced in both the CHO and S2 cell lines was properly processed, since we were unable to observe unprocessed FIX, which is expected to migrate at ~61 kDa.
The post-translational modification that is vital to the biological activity of FIX is the γ-carboxylation of 12 N-terminal glutamic acid residues, which is catalyzed by the γ-glutamyl carboxylase enzyme. Following demonstration of γ-carboxylase in Drosophila melanogaster (Li et al. 2000), in vitro studies showed that using equivalent amounts of Drosophila and human γ-carboxylase, the yield of γ-carboxylated products generated by the Drosophila γ-carboxylase was about 50 % higher as compared to that generated by the human enzyme (Bandyopadhyay et al. 2006). This apparent high activity of the Drosophila γ-carboxylase may be at the basis of the enhanced activity (35 %) and specific activity (20 %) observed here for S2-produced human FIX relative to that produced by CHO cells. Enrichment for γ-carboxylated FIX species via barium citrate adsorption further confirmed the presence of a functional γ-carboxylase in S2 cells, which indicated that up to 90 % of the FIX expressed by S2 cells was γ-carboxylated versus 79 % for the CHO cell line.
Drosophila S2 cells are also capable of N-terminal glycosylation of their protein products. Our data demonstrate that N-glycosylation of S2-expressed human FIX had occurred to a similar extent as in the FIX produced by the CHO cells. This was indicated by the generation of S2- and CHO-derived FIX products that both migrated at ~50 kDa when N-glycosylation pathways were blocked by tunicamycin. While the core structures of the N-glycans added by insect cells are known to be similar to those found in humans, the terminal structures have been reported to be less complex and lack sialic acid (Costa et al. 2014). FIX variants with lower sialic acid content may experience enhanced clearance (Blasko et al. 2013) and furthermore, non-human glycan structures have been suggested to modulate immunogenicity to the protein product (Costa et al. 2014; Dumont et al. 2015). Thus far, these issues have withheld the exploitation of Drosophila S2 cells for the production of any therapeutic protein.
The production of cost-effective and high-quality recombinant human FIX will have a marked impact on the treatment of hemophilia B patients worldwide. Our findings demonstrate that the S2 cell system has the potential of significantly reducing the cell development time, while maintaining FIX function. Future studies on this expression system are warranted in order to produce more humanized glycoproteins for therapeutic purposes. Overall, these observations support that the Drosophila S2 cell system is a good candidate for the efficient production of recombinant FIX.
References
Bandyopadhyay P, Clark K, Stevenson B, Rivier J, Olivera B, Golic K, Rong Y (2006) Biochemical characterization of Drosophilaγ-glutamyl carboxylase and its role in fly development. Insect Mol Biol 15:147–156
Blasko E, Brooks AR, Ho E, Wu JM, Zhao XY, Subramanyam B (2013) Hepatocyte clearance and pharmacokinetics of recombinant factor IX glycosylation variants. Biochem Biophys Res Commun 440:485–489
Bos MHA, van’t Veer C, Reitsma PH (2016) Molecular biology and biochemistry of the coagulation factors and pathways of hemostasis. Williams Hematology, 9th edn. McGraw-Hill, New York
Cherbas L, Cherbas P (2007) Transformation of drosophila cell lines: an alternative approach to exogenous protein expression. Methods Mol Cell Biol 338:317–340
Costa AR, Rodrigues ME, Henriques M, Oliveira R, Azeredo J (2014) Glycosylation: impact, control and improvement during therapeutic protein production. Crit Rev Biotec 34:281–299
Dumont J, Euwart D, Mei B, Estes S, Kshirsagar R (2015) Human cell lines for biopharmaceutical manufacturing: history, status, and future perspectives. Crit Rev Biotec. doi:10.3109/07388551.2015.1084266
Haddad-Mashadrizeh AA, Zomorodipour A, Izadpanah M, Sam MR, Sabouni F, Hosseini SJ (2009) A systematic study of the function of the human beta-globin introns on the expression of the human coagulation factor IX in cultured CHO cells. J Gene Med 10:941–950
Hansson K, Stenflo J (2005) Post-translational modifications in proteins involved in blood coagulation. J Thromb Haemost 3:2633–2648
Jallat S, Perraud F, Dalemans W, Balland A, Dieterle A, Faure T, Meulien P, Pavirani A (1990) Characterization of recombinant human factor IX expressed in transgenic mice and in derived trans-immortalized hepatic cell-lines. EMBO J 9:3295–3301
Jorge SA, Santos AS, Spina Â, Pereira CA (2008) Expression of the hepatitis B virus surface antigen in Drosophila S2 cells. Cytotechnology 57:51–59
Kim KR, Kim YK, Cha HJ (2008) Recombinant baculovirus-based multiple protein expression platform for drosophila S2 cell culture. J Biotechnol 133:116–122
Kim WH, Kim JS, Yoon Y, Lee GM (2009) Effect of Ca2+ and Mg2+ concentration in culture medium on the activation of recombinant factor IX produced in Chinese hamster ovary cells. J Biotechnol 142:275–278
Lee JM, Park JH, Park JO, Chang KH, Chung IS (2000) Expression of recombinant erythropoietin in stably transformed Drosophila melanogaster S2 cells. In Vitro Cell Dev Biol-Anim 36:348–350
Li T, Yang CT, Jin D, Stafford DW (2000) Identification of a drosophila vitamin K-dependent γ-glutamyl carboxylase. J Biol Chem 275:18291–18296
Liu J, Jonebring A, Hagström J, Nyström AC, Lövgren A (2014) Improved expression of recombinant human factor IX by co-expression of GGCX, VKOR and furin. Protein J 33:174–183
Moraes Â, Jorge SAC, Astray RM, Suazo CAT, Calderón CE, Augusto EFP (2012) Drosophila melanogaster S2 cells for expression of heterologous genes, From gene cloning to bioprocess development. Biotechnol adv 30:613–628
Nair A, Xie J, Hermiston TW (2011) Effect of different UCOE-promoter combinations in creation of engineered cell lines for the production of Factor VIII. BMC Res Notes 4:178
Nilsen SL, Castellino FJ (1999) Expression of human plasminogen in drosophila schneider S2 cells. Protein Expres Purif 16:136–143
Percival M, Bastien L, Griffin P, Kargman S, Ouellet M, O’Neill G (1997) Investigation of human cyclooxygenase-2 glycosylation heterogeneity and protein expression in insect and mammalian cell expression systems. Protein Express Purif 9:388–398
Thompson AR (1986) Structure, function, and molecular defects of factor IX. Blood 67:565–572
Vatandoost J, Zomorodipour A, Sadeghizadeh M, Aliyari R, Bos MH, Ataei F (2012) Expression of biologically-active human clotting factor IX in drosophila S2 cells: γ-carboxylation of a human vitamin K-dependent protein by the insect enzyme. Biotechnol Progr 28:45–51
Yao SN, Wilson JM, Nabel EG, Kurachi S, Hachiya HL, Kurachi K (1991) Expression of human factor IX in rat capillary endothelial cells—toward somatic gene therapy for hemophilia B. Proc Natl Acad Sci USA 88:8101–8105
Acknowledgments
This work has been supported by a grant (Project No. 372) from the Iran National Science Foundation (INSF) to Jafar Vatandoost.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Vatandoost, J., Bos, M.H.A. Efficient expression of functional human coagulation factor IX in stably-transfected Drosophila melanogaster S2 cells; comparison with the mammalian CHO system. Biotechnol Lett 38, 1691–1698 (2016). https://doi.org/10.1007/s10529-016-2156-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10529-016-2156-6