
Abstract
Objectives
To determine the role of endoplasmic reticulum (ER) stress and autophagy in apoptosis induced by bortezomib in human cervical cancer-derived HeLa cells and CaSki cells.
Results
Bortezomib treatment activated apoptosis, evidenced by increased expression of cleaved caspase-3 and cleaved PARP in both HeLa cells and CaSki cells. Bortezomib also induced the loss of the mitochondrial membrane potential, increased the level of ER stress-associated proteins GRP78, ATF4, and CCAAT-enhancer-binding protein homologous protein, and affected the expression of autophagy-related proteins; increasing the levels of LC3-II and ATG5–ATG12 and decreasing the level of p62. When we combined bortezomib with the ER stress activator tunicamycin, or autophagy inhibitors 3-methyladenine or chloroquine, cell growth inhibition and apoptosis were markedly enhanced.
Conclusions
Bortezomib activates apoptosis signaling, and activation of ER stress and inhibition of autophagy enhances the cytotoxicity of bortezomib, suggesting that these combination treatments may be potential chemotherapy strategies for treating cervical cancer.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The ubiquitin proteasome system (UPS) is the major pathway for protein degradation in cells. It is involved in many cellular processes, including degradation of abnormal or misfolded proteins targeted for destruction (Crawford et al. 2011). Dysregulation of protein degradation underlies many illnesses, including cancer. Bortezomib is a proteasome inhibitor that selectively blocks the activity of the 26S proteasome, and it is an effective agent against various types of solid cancers (Kiliccioglu et al. 2014). However, relapse following bortezomib therapy is common and results in multiple intractable tumors. Although the cellular mechanism of bortezomib resistance is not clear, it is known that cytotoxicity due to bortezomib can activate the intracellular defense response, such as endoplasmic reticulum (ER) stress and autophagy (Chauhan et al. 2005).
The ER plays an essential role in protein folding and secretion, calcium homeostasis, and lipid biosynthesis. Multiple endogenous and exogenous stimuli can interfere with ER function, causing unfolding and misfolding of proteins and leading to ER stress, which activates an evolutionarily conserved response known as the unfolded protein response (UPR). It can restore ER homeostasis by delivering misfolded proteins to the UPS (Szegezdi et al. 2006).
In addition to the proteasome degradation pathway, another mode of protein degradation is autophagy. Several studies have shown that autophagy has a pro-survival function by providing essential substrates for multiple cellular processes (Guo et al. 2011). However, the role of autophagy in cell death and drug resistance in cancer is still controversial. In this study, we evaluated the effect of bortezomib on apoptosis, autophagy, and ER stress in cervical cancer cells.
Materials and methods
Reagents
3-Methyladenine (3-MA), chloroquine (CQ), bortezomib, and tunicamycin were purchased from Sigma. Anti-LC3, anti-p62, anti-Atg5–Atg12, anti-caspase-3, anti-cleaved caspase-3, anti-poly (ADP-ribose) polymerase (PARP) and anti-tubulin were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA).
Cell culture
HeLa and CaSki cell lines were cultured in Iscove’s modified Dulbecco’s medium with 10 % (v/v) FBS. Cells were cultured in an environment of 5 % CO2 at 37 °C.
Cell viability assays
Viability of cells treated with bortezomib was measured with the MTT assay (Sigma). Briefly, cells were cultured in 96-well plates at 104 cells/well in 100 μl of complete medium. After treatment, MTT (20 μl, 5 mg/ml) was added to each well for another 4 h. Subsequently, DMSO (150 μl) was added to each well and the absorbance was read at 570 nm. Every group was repeated in six separate wells.
Mitochondrial membrane potential (MMP) analysis
JC-1 staining (Invitrogen Life Technologies) was used to detect the change in MMP by using flow cytometry. Briefly, cells were washed with cold PBS three times, and resuspended in PBS at 1.5 × 106 cells/ml and stained with 5 μl JC-1 (1 mg/ml) in the dark at 37 °C for 1 h. Finally, fluorescence was measured by FACS Calibur flow cytometer.
Western blot
After treatment, cells were washed twice with cold PBS and then 150 μl RIPA buffer was added to each group. Cell lysates were transferred to a 2 ml tube and shaken for 45 min in a cold room (4 °C). After harvesting the supernatant, total proteins (45 μg) were separated by 10 % (w/v) SDS-PAGE and proteins were transferred onto PVDF membranes. Then, the membranes were blocked in 5 % (v/v) skim milk for 2 h. Primary antibody was added and membranes were incubated in a cold room overnight. The next day, PVDF membranes were incubated with secondary antibody for 1 h at room temperature. The semi-quantitation of protein was done with a Tanon GIS gel imager system.
Statistical analysis
Data are representative of three independent experiments and were analyzed using one-way analysis of variance (Statistical Product and Service Solutions 11.5). P < 0.05 was considered significant.
Results
Bortezomib inhibits growth and induced apoptosis in human cervical cancer cell lines
HeLa and CaSki cells were treated with 0, 2.5, 5, 10, 20, and 50 nM bortezomib for 24 h. As shown in Fig. 1a, b bortezomib decreased the viability of HeLa cells and CaSki cells in a dose-dependent manner. We further measured the expression of apoptosis-related proteins in HeLa cells and CaSki cells treated dose-dependently with bortezomib (0, 5, 10 and 20 nM, for 24 h). The expression of cleaved caspase-3 and cleaved PARP was increased in both cell lines treated with bortezomib (Fig. 1c, f).

Bortezomib inhibits growth and induces apoptosis in human cervical cancer cells. a, b HeLa and CaSki cells were treated dose-dependently with bortezomib for 24 h. Cell viability was determined by MTT assay. Data are presented as mean ± SD, n = 6. *P < 0.05 versus control group; **P < 0.01 versus control group. c The expression of cleaved caspase-3 and cleaved PARP in HeLa cells treated dose-dependently with bortezomib for 24 h. d Quantitation of cleaved caspase-3 and cleaved PARP protein levels in HeLa cells. e The expression of cleaved caspase-3 and cleaved PARP in CaSki cells treated dose-dependently with bortezomib for 24 h. f Quantitation of cleaved caspase-3 and cleaved PARP protein levels in CaSki cells. Data are presented as mean ± SD, n = 3. *P < 0.05 versus control group
Bortezomib activates mitochondrial apoptosis signaling in human cervical cancer cell lines
Based on the results above, we also detected activation of the mitochondrial apoptotic pathway. HeLa cells and CaSki cells were dose-dependently treated with bortezomib (0, 5, 10 and 20 nM bortezomib for 24 h). As shown in Fig. 2a, the expression of cytoplasmic cytochrome c was increased significantly in HeLa cells treated with bortezomib. In addition, the Bcl-2 protein family was found to regulate the mitochondrial apoptotic pathway. Figure 2b shows that the ratio of Bax/Bcl-2 was increased in HeLa cells treated with bortezomib. We also measured MMP in HeLa cells treated with bortezomib. Bortezomib induced the loss of the MMP in HeLa cells (Fig. 2c). To confirm the results above, we repeated the experiment in CaSki cells, in which similar results were observed in CaSki cells treated with bortezomib (Fig. 2d–f).

Bortezomib activates mitochondrial apoptotic pathway in HeLa and CaSki cells. a The expression of cytochrome c in HeLa cells treated dose-dependently with bortezomib for 24 h detected by western blot. b The expression of Bax and Bcl-2 in HeLa cells treated dose-dependently with bortezomib for 24 h detected by western blot. Quantitation of the ratio of Bax/Bcl-2 in HeLa cells. c MMP was measured by JC-1 staining in HeLa cells. d The expression of cytochrome c in CaSki cells treated with bortezomib for 24 h. e The expression of Bax and Bcl-2 in HeLa cells treated dose-dependently with bortezomib for 24 h. Quantitation of the ratio of Bax/Bcl-2 in CaSki cells. f The MMP analysis was measured by JC-1 staining in CaSki cells. Data are presented as mean ± SD, n = 3. *P < 0.05 versus control group
Bortezomib initiates ER stress and induces ER stress-associated apoptosis in HeLa and CaSki cells
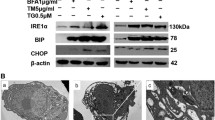
Besides the mitochondrial apoptotic pathway, ER stress-associated apoptosis is involved in apoptosis induced by anti-tumor agents. HeLa cells and CaSki cells were treated with 0, 5, 10 and 20 nM bortezomib for 24 h. In both cell lines the expression of GRP78 and ATF4 was increased, which indicates the presence of ER stress (Fig. 3). Additionally, we measured the expression of ER stress-associated apoptosis protein CCAAT-enhancer-binding protein homologous protein (CHOP). As shown in Fig. 3, the expression of CHOP was increased significantly in both HeLa cells and CaSki cells treated with bortezomib.

Bortezomib induces ER stress and ER stress-associated apoptosis in HeLa and CaSki cells. a Western blot analysis of GRP78, ATF4 and CHOP in HeLa cells treated dose-dependently with bortezomib for 24 h. b Quantitation of GRP78, ATF4, and CHOP protein levels in HeLa cells. c Western blot analysis of GRP78, ATF4 and CHOP in CaSki cells treated dose-dependently with bortezomib for 24 h. d Quantitation of GRP78, ATF4 and CHOP protein levels in CaSki cells. Data are presented as mean ± SD, n = 3, *P < 0.05 versus control group
Activation of ER stress by tunicamycin enhances growth inhibition and apoptosis induced by bortezomib in HeLa cells and CaSki cells
We pretreated cells with tunicamycin (5 mM) for 30 min as an ER stress activator to intensify the level of ER stress induced by bortezomib (10 nM, 24 h). MTT assay showed that bortezomib and tunicamycin enhanced the death of HeLa and CaSki cells treated by bortezomib (Fig. 4a, d). It also increased the expression of cleaved caspase-3 and cleaved PARP in HeLa and CaSki cells treated with bortezomib (Fig. 4b–f). These results indicate that upregulation of ER stress can enhance the sensitivity of HeLa cells and CaSki cells to bortezomib.

Activation of ER stress by tunicamycin enhances growth inhibition and apoptosis induced by bortezomib in HeLa and CaSki cells. a HeLa cells were treated with DMSO (Control) or pretreated with tunicamycin (5 mM, 30 min) with or without bortezomib (10 nM) for 24 h. Cell viability was determined by MTT assay. HeLa cells were treated as mentioned above. b The expression of cleaved caspase-3 and cleaved PARP were detected by western blot. c Quantitation of cleaved caspase-3 and cleaved PARP proteins level in HeLa cells. CaSki cells were treated as mentioned above. d Cell viability was determined by MTT assay. e The expression of cleaved caspase-3 and cleaved PARP were detected by western blot. f Quantitation of cleaved caspase-3 and cleaved PARP protein levels in CaSki cells. Data are presented as mean ± SD, n = 3. *P < 0.05 versus control group. #P < 0.05 versus bortezomib group
Bortezomib induces autophagy in HeLa cells and CaSki cells
We next measured the autophagy-related proteins in HeLa and CaSki cells. As shown in Fig. 5a, b bortezomib treatment (0, 5, 10 and 20 nM, for 24 h) increased the expression of LC3-II and ATG5–ATG12, and decreased the expression of p62 in HeLa cells. Similar results were observed in CaSki cells treated with bortezomib at the same doses and time as HeLa cells (Fig. 5c, d). These results indicate that bortezomib can induce autophagy in HeLa and CaSki cells.

Bortezomib-induced autophagy in HeLa and CaSki cells. a Western blot analysis for the expression of LC3, p62 and Atg5-Atg12 in HeLa cells treated dose-dependently with bortezomib for 24 h. b Quantitation of LC3-II, p62 and Atg5-Atg12 protein levels in HeLa cells. c Western blot analysis for the expression of LC3, p62 and Atg5-Atg12 in CaSki cells treated dose-dependently with bortezomib for 24 h. d Quantitation of LC3, p62 and Atg5-Atg12 protein levels in CaSki cells. Data are presented as mean ± SD, n = 3, *P < 0.05 versus control group
Inhibition of autophagy enhances growth inhibition and apoptosis induced by bortezomib in HeLa and CaSki cells
The exact effect of autophagy in bortezomib-induced apoptosis is unclear, so we used two different autophagy inhibitors (3-MA and CQ) to explore the effect of autophagy in bortezomib-induced apoptosis in cervical cancer cells. Both 3-MA (5 mM, 30 min pretreatment) and CQ (10 mM, 30 min pretreatment) (Fig. 6) further increased growth inhibition and apoptosis induced by bortezomib (10 nM for 24 h) in HeLa cells. The results were also confirmed in CaSki cells (Fig. 7).

Inhibition of autophagy by 3-MA and CQ enhances growth inhibition and apoptosis induced by bortezomib in HeLa cells. HeLa cells were treated with DMSO (control) or pretreated with 3-MA (5 mM, 30 min) with or without bortezomib (10 nM) for 24 h. a Cell viability was determined by MTT assay. b The expression of cleaved caspase-3 and cleaved PARP were detected by western blot. c Quantitation of cleaved caspase-3 and cleaved PARP protein levels in HeLa cells. HeLa cells were treated with DMSO (control) or pretreated with CQ (10 mM, 30 min) with or without bortezomib (10 nM) for 24 h. d Cell viability was determined by MTT assay. e The expression of cleaved caspase-3 and cleaved PARP were detected by western blot. f Quantitation of cleaved caspase-3 and cleaved PARP proteins in HeLa cells. Data are presented as mean ± SD, n = 6. *P < 0.05 versus control group. #P < 0.05 versus bortezomib group

Inhibition of autophagy by 3-MA and CQ enhances growth inhibition and apoptosis induced by bortezomib in Caski cells. Caski cells were treated with DMSO (control) or pretreated with 3-MA (5 mM, 30 min pretreated) with or without bortezomib (10 nM) for 24 h. a Cell viability was determined by MTT assay. b The expression of cleaved caspase-3 and cleaved PARP were detected by western blot. c Quantitation of cleaved caspase-3 and cleaved PARP proteins levels in Caski cells. Caski cells were treated with DMSO (control) or pretreated with CQ (10 mM, 30 min pretreated) with or without bortezomib (10 nM) for 24 h. d Cell viability was determined by MTT assay. e The expression of cleaved caspase-3 and cleaved PARP were detected by western blot. f Quantitation of cleaved caspase-3 and cleaved PARP proteins level in Caski cells. Data are presented as mean ± SD, n = 6. *P < 0.05 versus control group. #P < 0.05 versus bortezomib group
Activation of ER stress and inhibition of autophagy enhances growth inhibition induced by bortezomib
Finally, we treated HeLa cells with bortezomib (0 5, 10, and 20 nM, for 24 h), with and without tunicamycin (5 mM, 30 min pretreatment) and 3-MA (5 mM, 30 min pretreatment). The result of the MTT assay showed that the combination of the three agents further increased the sensitivity of HeLa cells to bortezomib significantly (Fig. 8a), indicating that in addition to activating the mitochondrial apoptosis pathway, bortezomib also induced ER stress and autophagy in cervical cancer cells. ER stress could further activate ER stress-associated apoptosis, which contributed to apoptosis induced by bortezomib. However, autophagy prevented bortezomib-induced apoptosis in cervical cancer cells (Fig. 8b). Thus, the combination of the ER stress activator, autophagy inhibitor, and bortezomib may provide a novel strategy for cervical cancer treatment.

Activation of ER stress and inhibition of autophagy enhances growth inhibition induced by bortezomib in HeLa cells. a HeLa cells were treated dose-dependently with bortezomib and pretreated with or without 3-MA (5 mM, 30 min) and tunicamycin (5 mM, 30 min) for 24 h. Cell viability was determined by MTT assay. b Bortezomib inhibits UPS activated by various biological processes to prevent or promote cell death predominantly by apoptosis. Activation of ER stress and inhibition of autophagy enhanced apoptosis induced by bortezomib. Data are presented as mean ± SD, n = 3. *P < 0.05 versus bortezomib group
Discussion
There is a general consensus that anti-tumor agents can induce the caspase-dependent mitochondrial apoptosis pathway, which activates a downstream cascade reaction, including apoptosome formation, and PARP and caspase-3 cleavage (Zeng et al. 2010). Our results indicate that bortezomib reduces cell viability in CaSki and Hela cells in a dose-dependent manner. Consistent with these results, the expression of cleaved PARP and cleaved caspase-3 were increased in both cell types, suggesting that bortezomib can induce apoptosis in cervical carcinoma cells. The MMP plays an important role in maintaining mitochondrial morphology and function, and mitochondrial dysfunction caused by alteration of MMP might be one of the early events in apoptosis. Moreover, the Bcl-2 protein family functions as a key regulator in this cell death pathway, especially Bcl-2 and Bax (Wei et al. 2007). Our results in CaSki and Hela cells treated with bortezomib indicate that the MMP was disrupted. In addition, bortezomib activated the intrinsic apoptotic pathway by stimulating cytochrome c release from mitochondria and increased the ratio of Bax/Bcl-2 in CaSki and Hela cells.
Increasing evidence demonstrates that, in addition to mitochondrial apoptosis, anti-tumor agents can induce apoptosis by up-regulating ER stress-associated apoptosis protein expression, such as CHOP (Zhong et al. 2012). Bortezomib can induce ER-associated apoptotic cell death in myeloma cells (Moriya et al. 2013). To investigate the relationship between ER stress and apoptosis induced by bortezomib in human cervical carcinoma, we detected the expression of ER stress-related proteins in human cervical carcinoma cells treated with bortezomib. Our data show that ER stress proteins, GRP78 and ATF4, were up-regulated and the expression of CHOP was increased in cells treated with bortezomib. The results demonstrate that both the mitochondrial dependent apoptosis pathway and ER stress dependent apoptosis participate in apoptosis induced by bortezomib. Our evidence also demonstrates that combination treatment with tunicamycin can further reduce cell viability of CaSki and Hela cells and increase the expression of cleaved caspase-3 and cleaved PARP.
Numerous anti-tumor agents can activate autophagy, and targeting autophagy may be a potential method in oncotherapy and drug resistance. Blocking autophagy may increase the effect of anti-tumor agents. Our results indicate that bortezomib can enhance the expression of autophagy-associated proteins LC3-II, p62 and Atg5–Atg12 complex, suggesting the occurrence of autophagy. Blocking autophagy by inhibitors 3-MA and CQ can increase the cytotoxicity of bortezomib. Moreover, we found that combination treatment with autophagy inhibitors can promote the expression of cleaved caspase-3 and cleaved PARP. These results suggest that autophagy is involved in apoptosis induced by bortezomib through a pro-survival mechanism.
In summary, our results show that in addition to mitochondrial dependent apoptosis, ER stress plays a key role in bortezomib-induced apoptosis by activating the down-stream ER stress-associated apoptosis pathway. Conversely, autophagy is characterized by a pro-survival effect which provides a novel target for tumor therapy. Hence, ER stress and autophagy both play an important role in apoptosis triggered by bortezomib, and in combination with other cancer drugs, it may provide a novel strategy for cancer treatment.
References
Chauhan D, Hideshima T, Anderson KC (2005) Proteasome inhibition in multiple myeloma: therapeutic implication. Annu Rev Pharmacol Toxicol 45:465–476
Crawford LJ, Walker B, Irvine AE (2011) Proteasome inhibitors in cancer therapy. J Cell Commun Signal 5:101–110
Guo JY, Chen HY, Mathew R, Fan J, Strohecker AM, Karsli-Uzunbas G, Kamphorst JJ, Chen G, Lemons JM, Karantza V, Coller HA, Dipaola RS, Gelinas C, Rabinowitz JD, White E (2011) Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev 25:460–470
Kiliccioglu I, Konac E, Varol N, Gurocak S, Yucel Bilen C (2014) Apoptotic effects of proteasome and histone deacetylase inhibitors in prostate cancer cell lines. Genet Mol Res 13:3721–3731
Kim I, Xu W, Reed JC (2008) Cell death and endoplasmic reticulum stress disease relevance and therapeutic opportunities. Nat Rev Drug Discov 7:1013–1030
Laussmann MA, Passante E, Düssmann H, Rauen JA, Würstle ML, Delgado ME, Devocelle M, Prehn JH, Rehm M (2011) Proteasome inhibition can induce an autophagy-dependent apical activation of caspase-8. Cell Death Differ 18:1584–1597
Moriya S, Che XF, Komatsu S, Abe A, Kawaguchi T, Gotoh A, Inazu M, Tomoda A, Miyazawa K (2013) Macrolide antibiotics block autophagy flux and sensitize to bortezomib via endoplasmic reticulum stress-mediated CHOP induction in myeloma cells. Int J Oncol 42:1541–1550
Szegezdi E, Logue SE, Gorman AM, Samali A (2006) Mediators of endoplasmic reticulum stress-induced apoptosis. EMBO Rep 7:880–885
Wei Q, Dong G, Franklin J, Dong Z (2007) The pathological role of Bax in cisplatin nephrotoxicity. Kidney Int 72:53–62
Zeng H, Zhang S, Yang KY, Wang T, Hu JL, Huang LL, Wu G (2010) Knockdown of second mitochondria-derived activator of caspase expression by RNAi enhances growth and cisplatin resistance of human lung cancer cells. Cancer Biother Radiopharm 25:705–712
Zhong JT, Xu Y, Yi HW, Su J, Yu HM, Xiang XY, Li XN, Zhang ZC, Sun LK (2012) The BH3 mimetic S1 induces autophagy through ER stress and disruption of Bcl-2/Beclin 1 interaction in human glioma U251 cells. Cancer Lett 323:180–187
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Zhang, Y., Bai, C., Lu, D. et al. Endoplasmic reticulum stress and autophagy participate in apoptosis induced by bortezomib in cervical cancer cells. Biotechnol Lett 38, 357–365 (2016). https://doi.org/10.1007/s10529-015-1968-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10529-015-1968-0