
Abstract
The activity of icarrin (a flavonoid from Herba epimedii) was investigated in the regulation of bone remodeling, a process coupled by osteoblast-mediated bone forming and osteoclast-mediated bone resorption. By directly co-culturing mouse bone marrow stromal cells and mouse preosteoclastic RAW264.7, and transwell co-culturing rat ovarian follicular granulosa cells (FGC), a 30 % increase in alkaline phosphatase (ALP) activity and 25 % increase in estradiol level occurred. Compared with the antiresorptive drug, alendronate, and an anabolic drug, PTH1–34, icarrin possessed all of the positive effects on the co-culture by increasing ALP activity, estradiol production and decreasing tartrate-resistant acid phosphatase activity. A similar action of icarrin occurred on co-culture of mesenchymal stem cells, mouse peripheral blood mononuclear cells, and FGC. Overall, by using a co-cultured cell-based in vitro screening assay, icarrin is suggested as a new class of dual-action therapeutic agent for osteoporosis.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Bone remodeling requires the coupling of osteoclast-mediated bone resorption and osteoblast-mediated bone formation in addition to hormone activity. Osteoclast differentiation from hematopoietic precursor cells depends on contact with osteoblast precursors or osteoblasts because these cells express osteoclastogenic cytokines such as RANKL (receptor for activator of nuclear factor-κB ligand) (Khosia 2001; Martin and Sims 2005). Osteoclasts also direct bone marrow mesenchymal stem cells (MSC) differentiation into osteoblasts through producing osteoblast-differentiating factors (Pederson et al. 2009). Interaction through cell–cell contact or through secreted coupling factors is thus needed for bone remodeling balance. Research is currently focusing on mimicking bone remodeling with the establishment of in vitro co-culture of osteoblasts with osteoclasts or osteoclast progenitors in splenic tissues (Burmeister et al. 2003), and then co-culture of osteoblasts and osteoclasts derived by human bone marrow stromal cells (BMSC) and monocytes (Heinemann et al. 2011; Fong et al. 2008).
Although two classes of drugs, including antiresorptive agents and anabolic agents, are available in clinics for treatment of osteoporosis, both of the therapies have limitations. They stimulate or inhibit both processes at the same time, which makes the imbalance of bone remodeling lasted. Research is currently focusing on drugs that regulate bone remodeling balance by inhibiting osteoclast-mediated bone resorption and osteoblast-mediated bone formation synchronously (Baron 2011; Zaidi and Iqbal 2012). Icarrin is a major flavonoid found in Herba epimedii, which possess beneficial effect on bone mass. Studies using cultured rat calvarial osteoblasts (Ma et al. 2011) or rat BMSC (Bian et al. 2011) showed that icarrin stimulated bone formation. In vitro study revealed that icarrin inhibited osteoclast differentiation and activity (Huang et al. 2007). However, whether icarrin could synchronously regulate both processes is still unclear.
We describe here the development of a cell-based model for screening and evaluating compounds that interfere with bone remodeling balance. To accomplish this, we created a co-culture system based on direct contact between MSC and preosteoclastic RAW264.7 or peripheral blood mononuclear cells (PBMC), and indirect contact with ovarian follicular granulosa cells (GC). Such a co-culture system supports simultaneous observation of activity of interacted cells, recapitulating the process of bone remodeling from osteoprogenitor differentiation to activation. By using this co-culture, we identified that icarrin inhibited osteoclast differentiation, increased MSC differentiation into osteoblasts and stimulated estrogen release synchronously.
Materials and methods
Reagents
Sodium alendronate was purchased from National Institute for Food and Drug Control, Beijing, China. Human recombinant parathyroid hormone1–34 (PTH1–34) was purchased from Prospec-Tany Technogene, Israel. Icarrin was isolated from Herba epimedii in our laboratory. Its purity was above 98 %. Other materials were obtained from Sigma-Aldrich unless otherwise stated.
Primary bone marrow mesenchymal stem cells (MSC)
BM-MSC was isolated according to a previously published protocol with some modification (Heinemann et al. 2011). Briefly, MSC were isolated from bone marrow, which aspirated from 8-week old BALB/c mice. MSC were collected using gradient centrifugation of stem cell-specific gradient solutions (Tianjin Haoyang Biological Manufacture Co. Ltd China). A layer of PBS buffered bone marrow cell fraction was placed on the top of gradient solution and centrifuged at 600×g for 20 min. The MSC fraction was collected and washed with PBS. MSC samples were resuspended in Minimum Essential Medium Alpha Medium (α-MEM, Gibco), supplemented with 20 % (v/v) fetal calf serum (FCS), 100 U penicillin/ml and 100 μg streptomycin/ml, and maintained at 37 °C with 5 % CO2 in a humidified atmosphere. On day 3, the MSC suspension was decanted and it was replaced with fresh complete medium. Upon 6–7 days culture, 90 % cell confluence was reached. These cell samples were employed with each experiment.
Primary peripheral blood mononuclear cells (PBMC)
PBMC were obtained from BALB/c mice and were isolated using gradient centrifugation of lymphocyte gradient solutions (Tianjin Haoyang) according to the manufacture’s instructions. PBMC were cultured in α-MEM, supplemented with 20 % FBS, 100 U/ml penicillin and 100 μg/ml streptomycin, and maintained at 37 °C with 5 % CO2 in a humidified atmosphere.
Primary ovarian follicular granulosa cells (GC)
Rat ovarian follicular GC were isolated from 4-week old female Sprague–Dawley rats 36 h after administration of 150 IU pregnant mare’s serum gonadotropin (PMSG) as described previously (Ireland et al. 2004). Briefly, ovaries were removed and placed in ice-cold PBS. Ovaries were cleaned of bursa cells and other extraneous tissues, and GC were collected from the surrounding media following follicle puncture. These cells were resuspended in Medium 199, supplemented with 20 % FBS, 100 U penicillin/ml and 100 μg streptomycin/ml, and maintained at 37 °C with 5 % CO2 in a humidified atmosphere. Upon 5 days culture, 90 % cell confluence was reached. These cell samples were employed with each experiment.
Direct co-culture of MSC and osteoclast precursor cells, and transwell co-culture of GC
Mesenchymal stem cells were plated at 5 × 104 cells/ml in 96-well plates, and cultured in α-MEM containing 20 % (v/v) FBS at 37 °C with 5 % CO2 atmosphere for 15 h. Osteogenic differentiation was induced by treatment with osteogenic supplement (OS) containing 100 nM dexamethasone, 1 mM β-glycerophosphate, and 5 μM l-ascorbic acid 2-phosphate for 8 days. Following this procedure, 5 × 103 RAW264.7 cells or PBMC were added to each BM-MSC seeded well and co-cultured for 6 days. Osteoclast differentiation was induced by treatment of RAW264.7 with 20 ng RANKL/ml or treatment of PBMC with 20 ng RANKL/ml plus 20 ng GM-CSF/ml for 4 days. Then 103 GC were added to transwell inserts positioned above the MSC and RAW264.7 confluent cell sheet and each well was maintained in α-MEM with free FBS. Co-cultures were maintained for another 2 days before analysis. Simultaneously, mono-culture of MSC, RAW264.7/PBMC, and GC was maintained as parallel control.
Double staining of alkaline phosphatase (ALP) and tartrate-resistant acid phosphatase (TRAP)
Cells were fixed with 60 % citrate-buffered acetone for 30 s. Then the cells were stained for ALP with 0.01 % naphthol AS-MX phosphate alkaline solution (pH 8.6) containing 0.3 mg Fast Blue RR/ml Salt according to the manufacturer’s instruction (Sigma). After the reaction solution was removed and discarded, the cells were washed with deionized water. The cells were further stained for TRAP with 0.1 M acetate solution (pH 5.0) containing 6.76 mM sodium tartrate, 0.12 mg naphthol AS-MX phosphate/ml, and 0.07 mg of fast Garnet GBC solution/ml as described in the manufacturer’s instruction (Sigma). Photomicrographs were obtained at 200× magnification.
Measurement of TRAP activity, ALP activity, and 17β-estradiol release
Estradiol production was determined by using an ELISA kit. After co-cultures were maintained for 2 days, 80 μl cell culture supernates and 20 μl GC lysis buffer were recovered for ELISA using rat estradiol ELISA Kit (Cusabio Biotech Co. Ltd, Wuhan, China) according to manufacturer’ instruction. Co-cultured BM-MSC and RAW264.7 cells were stained for ALP as described above. The ALP positive cells were enumerated by examination of the cultures under a microscope at 100× magnification. According to manufacturer’s instruction, ALP activity was semi-quantified by counting the number of ALP positive cells on the basis of quantity and intensity of precipitated dye within the cytoplasm of these cells. For measurement of TRAP activity, p-nitrophenyl phosphate (pNPP) was used as the substrate. Briefly, the fixed cells were washed with water for three times and were further incubated with 100 μl phosphatase substrate solution containing 10 mM pNPP and 10 mM sodium tartrate in 50 mM citrate buffer (pH 4.6) at 37 °C for 1 h. After incubation, the enzyme reaction mixture was transferred to another plate and the reaction was stopped with 100 μl 0.1 M NaOH. Absorbance at 405 nm was measured using an ELISA reader.
Drug assays
For the drug assays, each drug was added at different concentrations to the mono-culture or co-culture medium. MSC were exposed to PTH1–34 from the first 4 days, and then cultured in the absence of PTH1–34 during the remainder 4 days. When RAW264.7 cells were co-cultured, PTH1–34 was added again and cells were incubated until fixation at day 14 culture. For icarrin, the cells were exposed from day 1 to the end of the cultured period. Sodium alendronate was added from day 10 into RAW264.7 co-culture until fixation at day 15 culture. During the culture period, the culture medium was changed every 4 days. In the control group, the culture medium containing vehicle was also changed every 4 days. In an individual experiment, each experiment was tested in duplicate of triplicate wells.
The activities of TRAP, ALP, and estradiol release were examined on co-cultured or mono-cultured cells treated as for drug activity assays. For TRAP and ALP, the results were expressed as percentage of activity compared to OS plus RANKL-treated control. For estradiol release, the results were expressed as percentage of activity compared to untreated control on mono-culture or OS plus RANKL-treated control on co-cultures. The EC50 values were calculated according to the dose–response curve generated using a nonlinear regression sigmoidal dose–response curve (variable slope) by using GraphPad Prism 4.0 software (GraphPad Software Inc., USA).
Data analysis
Dose–response curves were calculated by non-linear regression analysis using GraphPad Prism software with the data previously normalized to the drug-untreated controls. All statistical analyses were performed using Student’s t test. Results were represented as the mean ± SEM. A p value of <0.05 was considered to be statistically significant in the tests.
Results and discussion
Functionally reciprocal impact occurs in MSC, RAW264.7 and GC co-cultures
MSC are the osteoprogenitors, which are differentiated toward mature osteoblastic phenotype in vitro in the presence of dexamethasone, β-glycerophosphate, and l-ascorbic acid 2-phosphate (Heinemann et al. 2011). The murine monocyte cell line RAW264.7, which can differentiate into osteoclast-like cells on the exposure to RANKL, represent a widely used in vitro model of osteoclast precursors.
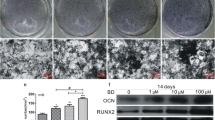
Upon 14 days MSC culture with OS modification, while 6 days co-culture with RAW264.7 cells and 2 days transwell co-culture with GC, the number of ALP staining-positive MSC as well as staining intensity increased, in contrast with mono-culture of MSC with OS modification (Fig. 1). When MSC were co-cultured with RAW264.7, the number of ALP staining-positive MSC increased from 21 ± 1/well to 25 ± 2/well. When RANKL was added in co-cultures, the number of ALP staining-positive MSC is 31 ± 2/well, a 50 % increase in ALP activity, suggesting that RANKL-induced osteoclastic RAW264.7 influence MSC differentiation. When GC were transwell co-cultured, the number of ALP staining-positive MSC reached 39 ± 2/well (Fig. 2a). This result suggests that GC influence MSC differentiation. Although MSC with OS modification also has the ability to induce osteoclastic differentiation without external addition of RANKL (Fig. 2b), the increase in TRAP activity is slight. To stimulate RAW264.7 to possess TRAP activity, higher enough to evaluate anti-resorptive effect of drugs, RANKL was added to the cultures in this study. RANKL elevated TRAP activity in RAW264.7 cells, a marker enzyme of osteoclasts. However, there is no significant difference between RAW264.7 mono-culture and co-cultures in the number of TRAP-positive cells (Fig. 1) or TRAP activity (Fig. 2b). In transwell assays, estradiol content from rat granulosa cells is 60 ± 2 pg/ml in the presence of MSC and RAW264.7 cells. When OS and RANKL were added to the co-culture, estradiol level reached 80 ± 15 pg/ml, indicating that differentiation of MSC and RAW264.7 influences estradiol production from GC (Fig. 2c).

MSC/RAW/GC co-culture shows more osteoblast differentiation. The altered activity of ALP and TRAP distinguished co-cultures from mono-culture of MSC and RAW264.7 after stimulation of MSC or RAW264.7 with OS and/or RANKL, respectively. The representative photomicrographs were made on cultures fixed on day 14. The ALP is stained with Naphthol AS-MX alkaline solution containing a diazonium salt (blue), while TRAP is labeled by Naphthol AS-BI in conjunction with a diazonium salt (purple to dark red)

Altered activity of ALP, TRAP, and estradiol production after co-culture of MSC, RAW264.7 and GC. The activities of ALP (a), TRAP (b) and estradiol production (c) at different stimulation case, including addition of RANKL or OS in mono-cultures or co-cultures, are presented on day 14 of the cultures. The data was representative of three independent experiments. *p < 0.05
Activity of icarrin was identified in co-culture
Next, we tested the response of the co-cultured cells to compounds including anti-resorptive drug alendronate, an anabolic drug PTH1–34, and the natural product icarrin. Having established the co-cultured form on day 14, at which time the activities of co-cultured cells are evident for TRAP (Park et al. 2004), ALP (Heinemann et al. 2011) and estradiol production (Ireland et al. 2004), we obtained the dose–response curve for these compounds following exposure to the compounds for the indicated days. TRAP activity from both of mono-cultured and co-cultured RAW264.7 cells were inhibited by alendronate, with similar EC50 values (11.9–8.64 μg/ml) (Fig. 3; Table 1). However, in the co-cultured MSC, ALP activity significantly decreased with exposure to alendronate higher than 2.5 μg/ml. PTH1–34 elevated ALP activity in both mono-cultures and co-cultures, but a marked increase in TRAP activity was also observed in the co-cultured case. While the co-culture form showed less lowered TRAP activity than the mono-culture form in response to icarrin, with a slight increase in the EC50 values (23.2–26.8 μg/ml), ALP activity in co-culture, displayed calculated EC50 value for icarrin (2.4 μg/ml) (Table 1). For estradiol production, alendronate and PTH1–34 had no effect either in co-culture or in mono-culture, but icarrin stimulated estradiol production from co-cultured GC.

Dose–response curve for the effect of PTH1–34, alendronate, and icarrin on TRAP activity, ALP activity, and estradiol production, observed in the co-cultures and mono-cultures. The co-cultured and mono-cultured cells were exposed to the varying compound concentrations and fixed on day 14. The curves obtained for the co-cultured form are presented in red, while those for the mono-cultured form are presented in black. The data was representative of two independent assays for alendronate and PTH1–34; and of three independent experiments for icarrin. For ALP activity, the number of ALP-positive MSC is 21 in mono-culture and 39 in co-culture as 100 % control; For TRAP activity, OD value is 0.42 in mono-culture and 0.46 in co-culture as 100 % control; For estrogen level, the concentration is 60 ng/ml in mono-culture and 80 ng/ml in co-culture as 100 % control. *p < 0.05 versus drug-treated mono-cultured group
In the co-cultured cell model, icarrin inhibited TRAP, stimulated ALP, and protected estradiol production in co-cultures, indicating that icarrin is a potential multi-target compound and has the ability of rebalancing bone remodeling. Although icarrin failed to significantly stimulate ALP activity from mono-cultured MSC, in co-cultures ALP activity was increased by icarrin. In parallel, estradiol level was elevated by icarrin in co-culture relative to mono-cultured GC, indicating that the co-cultured condition indeed impacts drug action. The concordance of these observations in vitro with the adverse effect of alendronate and PTH1–34 on osteoblastic ALP activity and osteoclastic TRAP activity respectively and thus resulted in osteosarcoma or bone loss in vivo (Rosen 2003; Whyte et al. 2003), suggesting that co-culture condition could be much closer to in vivo bone remodeling and therefore suitable for evaluation of compounds with bone remodeling-rebalancing activity.
To further develop a viable screening assay to identify icarrin activity, we modified the assay to replace RAW264.7 with primary PBMC before testing icarrin activity on the co-culture form. This was achieved by treating PBMC with 20 ng GM-CSF/ml and 20 ng RANKL/ml between day 8 and Day 14. Subsequent to this, most of PBMC present in the co-cultures became larger and TRAP staining-positive, as compared to untreated control (Fig. 4a). Icarrin at 5 μg/ml resulted in a decrease in TRAP staining tensity, and a simultaneous increase in the number of ALP staining-positive MSC. Also, estradiol production was increased by 30 % by 5 μg icarrin/ml.

Validating positive effect of icarrin on the co-cultures of MSC, PBMC and GC. Co-cultures were treated with or without OS and 5 μg/ml icarrin for 8 days, and then were left treated for a further 6 days with OS, RANKL, GM-CSF plus 5 μg icarrin/ml. The cultures were stopped with citrate-buffered acetone fixation on day 14. Double staining for ALP and TRAP was performed (a) and Estradiol production from supernate and GC lysis was examined by ELISA (b). The data was representative of three independent experiments, *p < 0.05
Conclusion
A first significant step towards the development of a cell-based assay aimed at the discovery of novel drugs that interfere with bone remodeling homeostasis is presented. Icarrin is identified as regulating bone remodeling from osteoprogenitor differentiation to activation and, especially, retains a sensitive estradiol environment in culture. Icarrin thus functions as a potent dual-action agent by synchronously increasing alkaline phosphatase activity, decreasing TRAP activity, and stimulating estradiol production. This study provides a molecular basis for the development of a combined anti-resorptive and anabolic agent capable of facilitating bone regeneration.
References
Bao JR, Yang JW, Li SF, Zhao W, Zhang Q, Yan Y (2005) Effects of icarrin on ovariectomized rats. Weisheng Yanjiu 34:191–193
Baron R (2011) Osteoporosis therapy-dawn of the post-bisphosphonate era. Nat Rev Endocrinol 8:76–78
Bian Q, Huang JH, Yang Z, Ning Y, Zhao YJ, Wang YJ, Shen ZY (2011) Effects of active ingredients in three kidney-tonifying Chinese herbal drugs on gene expression profile of bone marrow stromal cells from a rat model of corticosterone-induced osteoporosis. Zhong Xi Yi Jie He Xue Bao 9:179–185
Burmeister B, Domaschke H, Gelinsky M, Rösen-Wolff A, Hanke Th et al (2003) Co-culture of osteoblasts and osteoclasts on resorbable mineralised collagen scaffolds: establishment of an in vitro model of bone remodeling. Eur Cells Mater 5(suppl 2):18–19
Deal C (2009) Potential new drug targets for osteoporosis. Nat Clin Pract Rheumatol 5:20–27
Fong JE, Cassir N, Nihouannen DL, Komarova SV (2008) The role of osteoclasts in Osteoblast regulation. Eur Cells Mater 16(Suppl 4):22
Heinemann C, Heinemann S, Worch H, Hanke T (2011) Development of an osteoblast/osteoclast co-culture derived by human bone marrow stromal cells and human monocytes for biomaterials testing. Eur Cells Mater 21:80–93
Huang J, Yuan L, Wang X, Zhang TL, Wang K (2007) Icaritin and its glycosides enhance osteoblastic, but suppress osteoclastic, differentiation and activity in vitro. Life Sci 81:832–840
Ireland JL, Jimenez-Krassel F, Winn ME, Burns DS, Ireland JJ (2004) Evidence for autocrine or paracrine roles of alpha 2-macroglobulin in regulation of estradiol production by granulose cells and development of dominant follicles. Endocrinology 145:2784–2794
Khosia S (2001) Minireview: the OPG/RANKL/RANK system. Endocrinology 142:5050–5055
Ma HP, Ming LG, Ge BF, Zhai YK, Song P, Xian CJ, Chen KM (2011) Icarrin is more potent than genistein in promoting osteoblast differentiation and mineralization in vitro. J Cell Biochem 112:916–923
Martin TJ, Sims NA (2005) Osteoclast-derived activity in the coupling of bone formation to resorption. Trends Mol Med 11:76–81
Park EK, Kim MS, Lee SH, Kim KH, Park JY, Kim TH, Lee IS, Woo JT, Jung JC, Shin HI, Choi JY, Kim SY (2004) Furosin, an ellagitannin, suppresses RANKL-induced osteoclast differentiation and function through inhibition of MAP kinase activation and actin ring formation. Biochem Biophys Res Commun 325:1472–1480
Pederson L, Ruan M, Westendorf JJ, Khosla S, Oursler MJ (2009) Regulation of bone formation by osteoclasts involves Wnt/BMP signaling and the chemokine sphingosine-1-phosphate. Proc Natl Acad Sci USA 105:20764–20769
Rosen CJ (2003) The cellular and clinical parameters of anabolic therapy for osteoporosis. Crit Rev Eukaryot Gene Expr 13:25–38
Whyte MP, Wenkert D, Clemens KL, McAllister WH, Mumm S (2003) Bisphosphonate-induced osteopetrosis. N Engl J Med 349:457–463
Zaidi M, Iqbal J (2012) Double protection for weakened bones. Nature 485:47–48
Acknowledgments
This work was financially supported by the projects of National Natural Science Foundation of China (No. 81102739).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Liu, YQ., Han, XF., Liu, T. et al. A cell-based model of bone remodeling for identifying activity of icarrin in the treatment of osteoporosis. Biotechnol Lett 37, 219–226 (2015). https://doi.org/10.1007/s10529-014-1661-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10529-014-1661-8