
Abstract
Many γ-butyrolactone-autoregulator receptors control the production of secondary metabolites in Streptomyces spp. Hence, AvaR1, an autoregulator receptor protein in Streptomyces avermitilis, was characterized as a negative regulator of avermectin (Ave) production. Deletion of AvaR1 in a high-producing strain increased production of Ave B1a approx. 1.75 times (~700 μg/ml) compared with the parent strain. Semi-quantitative RT-PCR and electrophoretic mobility shift assays revealed that AvaR1 regulates the biosynthesis of Ave but not through the aveR pathway-specific regulatory gene. A special signaling molecule, avenolide, increased production of Ave. This study has refined our understanding of how avenolide regulates the production of Aves which is promising for developing new methods to improve the production of antibiotics in industrial strains.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Avermectin (Ave) and its analogues are well-studied anti-parasitic and insecticidal agents produced by Streptomyces avermitilis. They are composed of a 16-membered macrocyclic lactone with a disaccharide-methylated deoxysugar, l-oleandrose. Among the eight major active compounds, the B1a derivative has the most effective anti-parasitic activity (Fig. 1) (Burg et al. 1979; Egerton et al. 1979). The biosynthetic pathway of the Ave gene cluster has been elucidated and engineered to increase Ave production and develop new Ave analogues (Egerton et al. 1979; Ikeda and Omura 1997; Ikeda et al. 1999; Burg et al. 1979). In addition, the regulatory networks of Ave have been investigated. Specifically, aveR is a positive pathway-specific regulatory gene located within the Ave gene cluster (Ikeda et al. 1999; Guo et al. 2010; Kitani et al. 2009). Moreover, regulators located outside the gene cluster also play a critical role in the biosynthesis of Ave, such as the negative regulatory gene aveI (Chen et al. 2008) and the positive regulatory gene SAV 4023 (Duong et al. 2009). In these studies, the regulators affect Ave production in low producing strains. To improve the production of Ave further and its analogues in high producing strains, modification of regulatory networks could be a potential strategy (Alper et al. 2006; Nevoigt et al. 2007; Stephanopoulos 2007).

The structure of avermectins
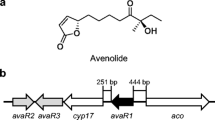
A large number of regulatory pathways control the production of secondary metabolites and morphological differentiation (Bibb 2005). The most well-known regulatory system in Streptomyces is the γ-butyrolactone autoregulator signaling cassette. This cassette uses γ-butyrolactone as an autoregulator to bind to a receptor protein, leading to the dissociation of the receptor from bound DNA and the expression of the target gene (Takano 2006; Horinouchi 2002; Ohnishi et al. 2005). Recently, a γ-butyrolactone autoregulator receptor protein, AvaR3, which is encoded by a gene outside of the Ave biosynthetic gene cluster, positively regulates the production of Ave (Miyamoto et al. 2011). For S. avermitilis, there are two other potential autoregulator receptor genes (avaR1 and avaR2) located in the vicinity of avaR3. Kitani et al. (2011) elucidated the structure of the AvaR1-interactive ligand, avenolide, which is a signaling molecule that positively regulates Ave-production. In this study, the role of AvaR1 in the regulation of Ave production was characterized in a high Ave producing strain, S. avermitilis M0 (supplementary data). Our results show that deletion of the avaR1 gene increases the production of Ave 1.75-times compared with the parent strain, suggesting that AvaR1 negatively regulates Ave production. Further investigation showed that AvaR1 indirectly down regulates Ave production and avenolide regulates Ave production through binding to AvaR1.
Materials and methods
Bacterial strains, plasmids, and medium
All bacterial strains and plasmids used in this study are listed in Supplementary Table 1. Primers are listed in Supplementary Table 2. Bacterial information, medium and culture conditions are shown in Supplementary data.
Construction of the avaR1-deletion mutant
The recombinant plasmid pTG2003 was constructed by in-frame deletion of the region encoding AvaR1. The 5′-flanking region containing the 1.6-kb external fragment of avaR1 was amplified with oligonucleotide primers 1 and 2. This PCR product was digested by HindIII and XbaI, and then inserted into the HindIII and XbaI sites of pGEM-3zf to generate pWJb216. The 3′-flanking DNA containing the 1.6-kb 3′-region of avaR1 was obtained by using primers 3 and 4. This PCR product was digested by XbaI and EcoRI, and inserted into the the XbaI and EcoRI sites of pGEM-3zf to generate pWJb182. The amplified PCR products were confirmed by DNA sequencing. The plasmid pWJb216 was digested with HindIII and XbaI, pWJb182 was digested with XbaI and EcoRI, and then 1.6-kb HindIII–XbaI and 1.6-kb XbaI–EcoRI fragments were introduced into the HindIII and EcoRI sites of the vector pKC1139 to generate the avaR1-disruption plasmid pTG2003. The resultant plasmid was introduced by intergenic conjugation into S. avermitilis M0 from E. coli ET12567, and the target gene was deleted by homologous recombination (supplementary data). The genotypes of the candidates for avaR1-deletion were confirmed by PCR (Supplementary Fig. 1).
Complementation of the avaR1 mutants
The DNA sequence encoding AvaR1 with its SD sequence was amplified with oligonucleotide primers 5 and 6. The PCR product was inserted into the StuI site of pANT841 to produce the plasmid pWJb5901 (clone avaR1), which was confirmed by DNA sequencing. The plasmid pLL6214 was digested by HindIII and EcoRI to obtain an ermEp* promoter gene. Plasmid pWJb5901 was digested by HindIII and XbaI to obtain a 0.7-kb HindIII–XbaI avaR1 fragment. The 0.45-kb HindIII–EcoRI ermEp* promoter fragment together with the 0.7-kb HindIII–XbaI avaR1 fragment were inserted into the XbaI and EcoRI sites of the vector pSET152 to produce the complementary plasmid pTG2004. Plasmid pTG2004 was introduced into the avaR1-disruption mutant by conjugation.
Transcriptional analysis by semi-quantitative RT-PCR
RNA was isolated from strains grown in fermentation medium for 72 h. Mycelia were collected and quickly frozen in liquid N2. RNA was extracted using Trizol. DNase I-treated RNA was used as a template for reverse transcription (RT) at 42 °C with a TaKaRa PrimeScript RT reagent Kit. Semi-quantitative analyses of the aveA1, aveA3, aveR, aveC and aco transcripts, normalized to hrdB encoding the major vegetative sigma factor, were performed. PCR was performed with 1 μl RT reaction mixtures and 10 pmol of each primer. The primers used for each gene were as follows: A1F and A1R for aveA1, A3F and A3R for aveA3, aveRF and aveRR for aveR, aveCF and aveCR for aveC, acoF and acoR for aco (which ecodes a key enzyme catalyzing the biosynthesis of the AvaR1—interactive ligand avenolide), and hrdBF and hrdBR for hrdB. Typically, the PCR procedure consisted of incubation of the reaction mixture at 95 °C for 5 min, followed by 30 cycles at 95 °C for 1 min, 55 °C for 30 s, and 72 °C for 30 s, ending with one cycle at 72 °C for 5 min. PCR amplification products, 10 μl, were loaded onto a 1.2 % agarose gel with ethidium bromide incorporated. Reaction products were visualized by the BioRad Gel Doc XR+ systems. The results were recorded by a video camera connected to a computer-based data analysis system. Densitometry measurements were quantitated using the image Lab 3.0 Analyzer software. PCR performed without RT served as a control to indicate the absence of contaminating DNA in the RT reaction mixtures.
Expression and purification of AvaR1
To express AvaR1, primer 7 and 8 were used to amplify avaR1. The PCR products were inserted into the StuI site of pANT841 to produce the plasmid pWJb705 (containing avaR1), which was confirmed by DNA sequencing. The plasmid pWJb705 was digested by XhoI and NdeI, and then the XhoI–NdeI avaR1 fragment was inserted into the NdeI and XhoI sites of the vector pET28a to produce the AvaR1 expression vector pTG2009. The plasmid pTG2009 was introduced into the protein expression strain E. coli BL21 (DE3), and was incubated in Luria broth containing 25 μg kanamycin/ml at 37 °C overnight. The culture was diluted 500 times into 500 ml fresh medium and cultivation was continued until an OD600 of 0.6–0.8 was reached. IPTG was added to 100 μM to initiate protein synthesis, and the cultivation was continued at 18 °C for 24 h. The cells were harvested by centrifugation and washed with lysis buffer (50 mM NaH2PO4, 300 mM NaCl, 20 mM imidazole). The washed cells were suspended in lysis buffer and disrupted by sonication, and the cellular debris was removed by centrifugation. The His6-tagged AvaR1 was purified using Ni-NTA-agarose gel. Purified protein was detected by SDS-PAGE, and the concentration was estimated by the Bradford method.
Electrophoretic mobility shift assays
Electrophoretic mobility shift assays (EMSA) were performed similarly to Uguru et al. (2005). The Cy5-labeled primer 9 and 10 were used to amplify the upstream region of aveR (0.222 kb). The Cy5-labeled primer 11 and 12 were used to amplify the upstream region of aco (0.3 kb). For the binding assay, ~5–10 ng Cy5-labeled DNA was incubated with avaR1 at 25 °C for 20 min in a buffer containing 10 mM Tris/HCl (pH = 8.0), 25 mM KCl, 2.5 mM MgCl2, 1 mM dithiothreitol, and 10 % (v/v) glycerol in 20 μl. After incubation, complexes and free DNA were resolved on non-denaturing polyacrylamide gels containing 4.5 % (v/v) polyacrylamide and 0.056 % bisacrylamide with a running buffer (0.5 % TBE buffer), at 100 V voltage at 4 °C for 2 h. The gels were visualized with Fuji Film FLA-9000.
Results and discussion
Sequence alignment between AvaR1 and ScbR
The avaR1 gene was found by blasting the homologous protein of one typical autoregulator receptor protein ScbR (Yang et al. 2005) in the S. avermitilis genome, which was annotated as an autoregulator receptor. Sequence alignment of avaR1 with the known autoregulator receptor ScbR was performed using the biology workbench 3.2 (Supplementary Fig. 2) and the AvaR1 structure was analyzed according to the crystal structure of CprB (Natsume et al. 2004). Analysis revealed that the receptor contained a DNA-binding helix-turn-helix (HTH) domain at the N-terminus. This result suggested that AvaR1 may bind to a specific DNA sequence and function as a regulator.
Effects of avaR1-disruption on Ave B1a production in S. avermitilis
The effects of autoregulator receptor disruption on antibiotics production have been widely studied (Takano 2006) and, in most cases, deletion of a receptor causes over-production of the target antibiotic. We first investigated the effects of deleting avaR1 in S. avermitilis M0 (TG2003). Compared with the parent strain S. avermitilis M0, Ave production in the avaR1-deletion strain TG2003 was increased significantly (Supplementary Fig. 3). The most efficient component (B1a) produced by TG2003 showed a 1.75-fold increase compared with M0 (Table 1). In the avaR1-complemented strain S. avermitilis TG2004, the production of Ave reverted to the level of M0 (Table 1), whereas Ave production remained the same level as TG2003 in the control strain that we obtained by introduction of pSET152 into TG2003 (data unpublished). Therefore, all of the results verified that AvaR1 acts as a negative regulator of Ave biosynthesis. In some cases, autoregulator receptors control both the production of antibiotics and morphological differentiation (Takano 2006). We compared the morphology of S. avermitilis TG2003 with the M0 strain, but no apparent difference was observed in either MS or YEME culture medium.
RT-PCR analysis of the transcriptional level of Ave biosynthetic genes and related regulatory genes
To examine whether AvaR1 regulates the Ave genes directly and to elucidate the relationship between AvaR1 and the biosynthesis of the signaling molecule avenolide, semi-quantitative RT-PCR analysis was performed. The results are shown in Fig. 2. The mRNA levels of aveA1, aveR, aveA3 and aveC were all increased in TG2003 compared with M0. Since regulation of Ave production by AvaR1 may occur via inhibition of avenolide production, the mRNA level of aco, a key gene in the biosynthesis of avenolide was also measured. The mRNA level of aco did not show an apparent increase in TG2003, suggesting that AvaR1 regulates the biosynthesis of Ave possibly by acting on aveR expression or alternatively, by inhibiting some other genes related to Ave production.

Semi-quantitative RT-PCR analysis of the transcription-level of the PKS (AvaA1 and AvaA2), the pathway-specific regulator (AveR), an unknown function gene involved in biosynthesis Ave (aveC), and a key gene for biosynthesis the avenolide (aco) in M0 strain and the mutants (after fermented for 72 h)
EMSA analysis of the regulatory mechanism of AvaR1
To obtain further insight into the regulatory mechanism of AvaR1, EMSA was performed and the results are summarized in Fig. 3. By incubating Cy5-labeled aco promoter DNA with AvaR1, the band was observed to shift significantly (Fig. 3b) thus confirming the reported interaction between the aco promoter and AvaR1. To determine whether the promoter DNA of aveR reacted with AvaR1, a 10-times excess of unlabeled promoter DNA of aveR was added into the Cy5-labeled aco-AvaR1 complex for competition. As clearly shown in Fig. 3b, the shifted band remained with competing aveR promoter DNA in the system. In addition, no shifted band appeared after incubating Cy5-labeled aveR promoter DNA with AvaR1. As a control, a large excess of unlabeled aco promoter DNA was used compete with Cy5-labeled aco promoter DNA, and the shifted band disappeared completely, indicating that AvaR1 was binding to the promoter of aco specifically.

a Purification of His-tagged of AvaR1. Left lane 116 KD protein marker; right lane AvaR1. b Assay of DNA binding of AvaR1. Lane 1 12.5 ng free cy5-labeled aco promoter DNA, lane 2 0.5 μg AvaR1 with 12.5 ng cy5-labeled aco promoter DNA; lane 3 125 ng unlabeled aco promoter DNA with the reaction system in lane 2; lane 4 125 ng unlabeled aveR promoter DNA with the reaction system in lane2; lane 5 12.5 ng cy5-labeled aveR promoter DNA; lane 6, 0.5 μg AvaR1 with 12.5 ng cy5-labeled aveR promoter DNA
Taken together, these results suggest that AvaR1 does not regulate AveR directly; rather, it binds to the promoter of aco. Since AvaR1 had no effect on the expression of aco in the RT-PCR experiment, AvaR1 may regulate the production of Ave by inhibition of some genes that positively regulate the production of Ave. A large amount of avenolide can increase the production of Ave 1.8-fold compared with its absence in a fermentation culture (Kitani et al. 2011). The threshold of avenolide that regulates the production of Ave resulted in similar Ave production to the AvaR1-deletion mutants. These results suggest that avenolide possibly regulates the production of Ave by binding to AvaR1, stimulating protein release from target DNA to induce expression of the genes related to Ave production (Fig. 4).

The proposed regulatory mechanism of AvaR1. AvaR1 binding to several proposal sites. One is in the front of aco, the aco gene is involved in production of avenolide, and the others are in front of other genes which related to the production of avermectins. The avenolide (black balls) is always bind to AvaR1 (black squares) relieves the suppression
A special autoregulator receptor, ArpA, exists in Streptomyces that binds to the promoter of the transcriptional regulator gene, adpA, and inhibits the expression of a number of other genes (Ohnishi et al. 2005). Although there is also an adpA ortholog in S. avermitilis, named bdpA (SAV5023) (Zhu et al. 2005), under our culture conditions the transcription level of bdpA in S. avermitilis TG2003 was similar to that in S. avermitilis M0 (unpublished data, this laboratory). Therefore, AvaR1 should be a bdpA-independent regulator.
In conclusion, previous work has elucidated the function of the AvaR1-interactive ligand avenolide (Kitani et al. 2011), yet there were no reports about AvaR1. Here, we characterized AvaR1 in S. avermitilis and showed that avaR1 can remotely and indirectly regulate Ave biosynthesis. In addition, this suggests that avenolide regulates Ave production through binding to AvaR1. While it is difficult to increase the production of Ave by increasing the production of avenolide, our studies indicate that optimizing the regulator receptor could be a more straightforward and effective method to achieve this goal. There have been several previous studies on Ave production-related regulators but most of these were performed in low-yield strains. In this study, a high producing strain was engineered to improve production of Ave by 1.75-fold, providing a powerful strategy in industrial uses.
References
Alper H, Moxley J, Nevoigt E, Fink GR, Stephanopoulos G (2006) Engineering yeast transcription machinery for improved ethanol tolerance and production. Science 314:1565–1568
Bibb MJ (2005) Regulation of secondary metabolism in Streptomycetes. Curr Opin Microbiol 8:208–215
Burg RW, Miller BM, Baker EE, Birnbaum J, Currie SA, Hartman R, Kong YL, Monaghan RL, Olson G, Putter I, Tunac JB, Wallick H, Stapley EO, Oiwa R, Omura S (1979) Avermectins, new family of potent anthelmintic agents-producing organism and fermentation. Antimicrob Agents Chemother 15:361–367
Chen L, Lu YH, Chen J, Zhang WW, Shu D, Qin ZJ, Yang S, Jiang WH (2008) Characterization of a negative regulator AveI for avermectin biosynthesis in Streptomyces avermitilis NRRL8165. Appl Microbiol Biotechnol 80:277–286
Duong CTP, Lee HN, Choi SS, Lee SY, Kim ES (2009) Functional expression of SAV3818, a putative tetr-family transcriptional regulatory gene from Streptomyces avermitilis, stimulates antibiotic production in Streptomyces species. J Microbiol Biotechnol 19:136–139
Egerton JR, Ostlind DA, Blair LS, Eary CH, Suhayda D, Cifelli S, Riek RF, Campbell WC (1979) Avermectins, new family of potent anthelmintic agents-efficacy of the bia component. Antimicrob Agents Chemother 15:372–378
Horinouchi S (2002) A microbial hormone, A-factor, as a master switch for morphological differentiation and secondary metabolism in Streptomyces griseus. Front Biosci 7:D2045–D2057
Ikeda H, Omura S (1997) Avermectin biosynthesis. Chem Rev 97:2591–2609
Ikeda H, Nonomiya T, Usami M, Ohta T, Omura S (1999) Organization of the biosynthetic gene cluster for the polyketide anthelmintic macrolide avermectin in Streptomyces avermitilis. Proc Natl Acad Sci USA 96:9509–9514
Kitani S, Ikeda H, Sakamoto T, Noguchi S, Nihira T (2009) Characterization of a regulatory gene, aveR, for the biosynthesis of avermectin in Streptomyces avermitilis. Appl Microbiol Biotechnol 82:1089–1096
Kitani S, Miyamoto KT, Takamatsu S, Herawati E, Iguchi H, Nishitomi K, Uchida M, Nagamitsu T, Omura S, Ikeda H, Nihira T (2011) Avenolide, a Streptomyces hormone controlling antibiotic production in Streptomyces avermitilis. Proc Natl Acad Sci USA 108:16410–16415
Miyamoto KT, Kitani S, Komatsu M, Ikeda H, Nihira T (2011) The autoregulator receptor homologue AvaR3 plays a regulatory role in antibiotic production, mycelial aggregation and colony development of Streptomyces avermitilis. Microbiology 157:2266–2275
Natsume R, Ohnishi Y, Senda T, Horinouchi S (2004) Crystal structure of a γ-butyrolactone autoregulator receptor protein in Streptomyces coelicolor A3(2). J Mol Biol 336:409–419
Nevoigt E, Fischer C, Mucha O, Matthaeus F, Stahl U, Stephanopoulos G (2007) Engineering promoter regulation. Biotechnol Bioeng 96:550–558
Ohnishi Y, Yamazaki H, Kato JY, Tomono A, Horinouchi S (2005) AdpA, a central transcriptional regulator in the A-factor regulatory cascade that leads to morphological development and secondary metabolism in Streptomyces griseus. Biosci Biotechnol Biochem 69:431–439
Stephanopoulos G (2007) Challenges in engineering microbes for biofuels production. Science 315:801–804
Takano E (2006) γ-Butyrolactones: Streptomyces signalling molecules regulating antibiotic production and differentiation. Curr Opin Microbiol 9:287–294
Yang YH, Joo HS, Lee K, Liou KK, Lee HC, Sohng JK, Kim BG (2005) Novel method for detection of butanolides in Streptomyces coelicolor culture broth, using a His-tagged receptor (ScbR) and mass spectrometry. Appl Environ Microbiol 71:5050–5055
Zhu DQ, He XY, Zhou XF, Deng ZX (2005) Expression of the melC operon in several Streptomyces strains is positively regulated by AdpA, an AraC family transcriptional regulator involved in morphological development in Streptomyces coelicolor. J Bacteriol 187:3180–3187
Acknowledgments
We thank the grants from National Basic Research Program of China (973 Program) 2009CB118901 and the Chinese Academy of Science for the financial support. We thank the group of Prof. Weihong Jiang for providing the technical help.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Wang, JB., Zhang, F., Pu, JY. et al. Characterization of AvaR1, an autoregulator receptor that negatively controls avermectins production in a high avermectin-producing strain. Biotechnol Lett 36, 813–819 (2014). https://doi.org/10.1007/s10529-013-1416-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10529-013-1416-y