
Abstract
An integrative gene expression system has been constructed for the directional assembly of biological components in Synechocystis PCC6803. We have characterized 11 promoter parts with various expression efficiencies for genetic engineering of Synechocystis for the production of fatty alcohols. This was achieved by integrating several genetic modifications including the expression of multiple-copies of fatty acyl-CoA reductase (FAR) under the control of strong promoters, disruption of the competing pathways for poly-β-hydroxybutyrate and glycogen synthesis, and for peptide truncation of the FAR. In shake-flask cultures, the production of fatty alcohols was significantly improved with a yield of 761 ± 216 μg/g cell dry weight in Synechocystis, which is the highest reported to date.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Cyanobacteria have the potential for photosynthetic and sustainable production of high-value bio-products such as biofuels, biochemicals, drugs, natural nutrients from CO2 (Ducat et al. 2011). They can produce fatty alcohols and fatty alka(e)nes as biofuels (Tan et al. 2011). To improve the titers of desired products, it is crucial to modify the pathways systematically through metabolic engineering and synthetic biology strategies. Generally, the regulation of enzyme expression levels and the elimination of the competing pathways are essential to increase the metabolic flux towards desired metabolites (Boyle and Silver 2012). Hence, there is an urgent need to develop molecular tools and characterize the biosynthetic parts to modulate the metabolic pathways in cyanobacteria. Promoters are important regulatory elements that control protein expression from both transcriptional and translational levels. The various spacer regions between Shine–Dalgarno (SD) and the start codon (AUG) strongly influence the protein translation in both E. coli and cyanobacteria (Matteucci and Heyneker 1983; Takeshima et al. 1994). To date, numerous promoters such as Ptrc, Ptac, PrbcL, PpsbA2, and the copper-inducible promoter, PpetE, (Tan et al. 2011; Huang et al. 2010; Liu and Sheng 2011; Gao et al. 2012) have been applied for biofuel production in cyanobacteria. However, few of them have been evaluated for quantitative control of metabolic steps to maximize the product yields. Thus the screening and characterization of the promoter parts are significant for optimization of biofuel-producing pathways in cyanobacteria.
Synthetic biology enables us to re-design existing biological systems and characterize the biological parts for useful purposes. To develop molecular tools for metabolic engineering of cyanobacteria and other biotechnological applications, we constructed an integrative platform for directional assembly of synthetic parts containing homologous recombination fragments, promoter, target gene, and selection marker. This platform can be used for the efficient gene expression and the disruption of competing pathways. We also characterized a set of promoters to offer different expression efficiencies for target genes and constructed an agp (ADP-glucose pyrophosphorylase gene)-targeting plasmid to disrupt the glycogen synthesis. Furthermore, we successfully applied these molecular tools for optimizing the fatty alcohol production in Synechocystis sp. PCC6803.
Materials and methods
Materials
The kits used for molecular cloning were from Omega Bio-tek (USA) or Takara Biotechnology (Japan). Oligo nucleotides were carried out by Sangon (Shanghai, China). Taq DNA polymerases, DNA ladders and all restriction endonucleases were from Fermentas or Takara Biotechnology (Japan). The pentadecanol was purchased from Sigma-Aldrich. Other chemicals were from Merck or Ameresco.
Strains and growth conditions
E. coli DH5α was used for routine DNA transformation and plasmid isolation. E. coli strains were routinely grown in LB broth at 37 °C or on LB plates supplemented with 1.5 % (w/v) agar. 50 μg spectinomycin/ml, 50 μg kanamycin/ml or 34 μg chloramphenicol/ml was added when required.
Synechocystis sp. PCC6803 and other derived strains were cultured at 30 °C in BG11 medium (Stanier et al. 1971) under continuously light illumination (30–50 μmol/m2 s). 20 μg spectinomycin/ml, 20 μg kanamycin/ml or 20 μg erythromycin/ml was added when necessary. For enzymatic assay, the strains were cultured in 50 ml glass flasks with 20 ml BG11 liquid medium in an illuminated shaker at 140 rpm. OD730 values were used to determine biomass densities. For fatty alcohol analysis, the Synechocystis strains were grown in 500 ml flasks containing 300 ml medium bubbled with filtered air and harvested when approaching the stable phase.
Construction of the integrative vector pXT37a and promoter parts
All primers used are listed in Supplementary Table 1. All plasmids and strains are summarized in Supplementary Table 2. The pKW1188 (Williams 1988) derived plasmid pHB1536 and pHB1567 contain the homologous fragments of the slr0168 gene, the PpetE promoter, the spectinomycin-resistance gene and the reporter gene lacZ (Gao et al. 2007), etc. We re-assembled these synthetic parts, created single enzyme restriction sites flanking them, and obtained the integrative platform pXT37a (Supplementary Fig. 1). Promoters for the Synechocystis gene atpB (encoding ATP synthase beta subunit), psaD (encoding photosystem I subunit II), psbA1 (encoding photosystem II D1 protein), psbA2 (encoding photosystem II D1 protein), psbB (encoding photosystem II core light harvesting protein), and rbcL (encoding rubisco large subunit) were amplified from the genomic DNA of Synechocystis. They were also modified by mutating their spacer regions (Supplementary Fig. 3) using the designed primers. The lactose promoter Plac was amplified using pK18 as the template. The PCR products were firstly cloned into the cloning vector pMD18-T (Takara, Japan) and the sequences were confirmed by Sanger sequencing using the primer M13F-47 and M13R-48. Then the un-modified promoters were subsequently subcloned to BglII/KpnI site of the plasmid pXT37a, while the modified promoters were inserted into the NdeI/KpnI site to obtain the expression plasmids.
Expression of fatty acyl-CoA reductase in Synechocystis
The 3.2 kb fragment containing agp and its flanking regions was amplified using agp-1/agp-2 primers and inserted into pMD18-T vector, generating the plasmid pKC100 (Supplementary Fig. S4). The ClaI/SmaI fragment of pKC100, the C.K2 antibiotic marker (Elhai and Wolk 1988), the PpsbD13 promoter and the fatty acyl-CoA reductase (FAR) gene at3g11980 (Tan et al. 2011) were then assembled based on the expression platform pFQ34b and the agp targeting plasmid pLY21 was obtained. Similarly, the NcoI fragment pKC104 (Tan et al. 2013), the C.CE2 antibiotic marker (Elhai and Wolk 1988), the PrbcL12 promoter and the FAR gene far_jojoba (Tan et al. 2011) were orderly assembled based on the platform pFQ31a and the phaAB targeting plasmid pLY25 was obtained. The Ω-PpetE-at3g11980 fragment obtained from pXT34 was inserted into the XbaI and KpnI site replacing the Omega fragment in pXT51 (Tan et al. 2011) and the plasmid pLY10 was obtained. The 114 N-terminal amino acid residues of At3g11980 was truncated using the template pXT34 (Tan et al. 2011) and the primer at3g11980-trunc-1 and at3g11980-trunc-2. The resulted at3g11980-trunc gene was ligated to pFQ34b, generating the plasmid pLY43. All the PCR products were confirmed by sequencing and all the constructs were checked by enzyme digestion.
Strain transformation and genomic integration
The integrative plasmids were transformed to Synechocystis according to the established method (Williams 1988) with minor modifications (see Supplementary data). Through homologous recombination with the slr0168, agp or phaAB gene locus, the gene expression cassette in these plasmids was integrated into the genome of Synechocystis. Meanwhile, the gene slr0168, agp and phaAB were disrupted since their native parts in the genome were replaced by the recombined fragments. The double recombinants were selected on BG11 plates with corresponding antibiotics (Williams 1988). Homologous integration of the expressing cassette and segregation of all Synechocystis recombinants were verified by genomic PCR (Supplementary Fig. S2) using the candidate gene specific and slr0168/agp/phaAB specific primers. The confirmed Synechocystis recombinants were used for β-d-galactosidase assay and fatty alcohol analysis.
Enzyme assay for β-d-galactosidase
β-d-Galactosidase activity was determined according to a modified Miller test (Gao et al. 2007). Synechocystis cultures (OD730 = 1.2–1.5) were harvested and the OD730 was adjusted to 1.5. Then 1.5 ml cells were centrifuged and re-suspended in 1 ml Z buffer. 50 μl SDS (0.1 %, w/v), 50 μl chloroform and 0.2 ml ONPG was sequentially added to each sample, incubated at 30 °C for 20 min and finally 0.5 ml 1 M Na2CO3 was applied to stop the reaction. A420 value of the supernatant was recorded. A reaction without cells was used as a blank control. β-Galactosidase activity was calculated using the Eq. (1)
Fatty alcohol extraction and GC–MS analysis
Hundred milliliter Synechocystis cultures (OD730 = 4–5) were harvested and prepared for fatty alcohol analysis as described with minor modifications (Tan et al. 2011). The cells were suspended in 10 ml TE buffer (pH 8.0) and lysed by sonication, then the lysate was extracted with 10 ml chloroform/methanol (2:1, v/v) for 30 min. Prior to extraction, 20 μg 1-pentadecanol was added as the internal standard for alcohol quantification in each sample. The fatty alcohol contents in the mutants were quantified by GC–MS system equipped with a HP-INNOWax column (30 m × 250 mm × 0.25 mm). Helium was used as carrier gas. For fatty alcohol analysis, the injector was at 250 °C and the following program was applied: 100 °C for 1 min, increase of 5 °C/min to 200 °C, then increase of 25 °C/min to 240 °C and held for 15 min.
Results and discussion
Construction of the integrative vector of pXT37a for directional assembly of biosynthetic parts
The genomic integration strategy (Fig. 1A) offers stable gene expression and enables directional integration of synthetic parts into the specific locus of the genome via homologous recombination (Williams 1988). pXT37a (Fig. 1B) contains the PpetE promoter, the ribosome binding site (rbs) sequence between BglII and NdeI site (AGATCTGACTAACTGAGGAGGATTGCATATG), the reporter gene lacZ, the spectinomycin-resistance gene, the homologous recombination fragments for the slr0168 neutral site and a bacterial replicon. It has wide potential for heterologous gene expression and/or gene knock-out in Synechocystis. Its gene expression cassette can integrate into the slr0168 locus of the Synechocystis genome by homologous integration. Besides, it has restriction sites flanked the synthetic parts and allows: (1) assembly of promoter part by insertion into KpnI and BglII (no need an rbs) or KpnI/NdeI sites (an rbs should be added); (2) insertion of target genes using the upstream BglII/NdeI sites and the downstream EcoRI/XbaI/XhoI sites; (3) assembly of antibiotic-resistance gene or other selective markers into XbaI and KpnI site; (4) assembly of homologous recombination arms and bacterial replicon via insertion to XbaI/XhoI or the XbaI/XbaI site. Thus any synthetic part can be assembled into pXT37a by replacing the current fragments using the established cloning methods.

The utilization of genome integration strategy in cyanobacteria and the plasmid map of pXT37a. A The utilization of integration strategy by changing the elements in the integrated expression cassette. a The insertion of a heterologous gene into the ORF of the target locus. b The insertion of an expression cassette into a neutral site. c The knock-out of target gene by integration of a selection marker. d The disruption of target gene by insertion of a selection marker. ori The origin of replication; M antibiotic-resistance gene or other selection markers; L left arm for homologous recombination; R right arm for homologous recombination; P the promoter of the integration locus; P’ the heterologous promoter from the expression cassette; G the native gene at the integration locus; G’ the target gene from the expression cassette. B The integrative vector of pXT37a. ori The origin of replication; Ap r ampicillin-resistance gene sequence; PpetE the copper inducible promoter for petE gene
Design and characterization of promoters with various expression efficiencies
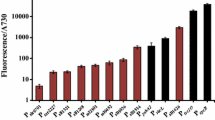
lacZ was selected to measure the expression behavior under the promoters. The β-d-galactosidase assay has high sensitivity and low background by avoiding the interference from photosynthetic pigments or the intracellular aldehydes that affect the reporters of fluorescent proteins (Schaefer and Golden 1989). Supplementary Table 2 lists the strains expressing lacZ gene under seven Synechocystis promoters (PatpB, PrbcL, PpsbB, PpsaD, PpsbD, PpsbA1, and PpetE), one bacterial promoter Plac and eleven modified Synechocystis promoters. Figure 2 shows the β-d-galactosidase activity in these strains.

β-Galactosidase activity in Synechocystis mutant strains after applying different promoters. The results graphed are averages derived from three independent experiments. The mean values of the β-galactosidase were plotted in the figure (error bars represent the standard deviation). β-Galactosidase activity in the Synechocystis strains are abbreviated with the name of the promoter that used in these strains. All the native promoters were named as Pgene-12 (e.g.: PrbcL-12); the modified promoters were named as Pgene-13, 14 or 15 (e.g.: PrbcL-13, PrbcL-14 or PrbcL-15); the strains Syn-LY2 containing a spectinomycin-resistance gene in the slr0168 site of the Synechocystis genome was used as the negative control. The strain Syn-HB1567 (Gao et al. 2007) that expresses lacZ gene under the PpetE promoter at the slr0168 site was used as the positive control. LY2 Syn-LY2; PpetE-1567 PpetE promoter in Syn-HB1567; PpetE-37a PpetE promoter in Syn-XT37a; PpetE-37a PpetE promoter in Syn-XT37b. The detailed strain information is summarized in Supplementary Table 2; sequences of the modified promoters are shown in Supplementary Fig. 3
PpetE produces a β-d-galactosidase activity around 60 Miller units when Cu2+ in the media is over 400 nM (Gao et al. 2007). In Fig. 2, the PpetE in Syn-HB1567 achieved a compared strength of 71.5 Miller units in BG11 media (Cu2+ at ~316 nM) in our experiment conditions. Syn-XT37a and Syn-XT37b exhibited similar strengths of PpetE, proving the feasibility to use pXT37a as the platform for promoters screening. The photosystem promoters of PrbcL, PpsbB, PpsaD and PpsbD all showed strong expression of over 95 Miller units. The PatpB promoter revealed the strongest expression (105 Miller units) among the native promoters. The psbA1 gene in Synechocystis is a silent and divergent copy of the psbA gene family but was activated by exchanging part of its upstream region with a corresponding fragment of the psbA2 copy. PpsbA1-12 was active and exhibited weak β-D-galactosidase expression (29.6 Miller units) applying the rbs from the pXT37a. However, the PpsbA1-13 with 2-bp deletion in the spacer region demonstrated a much lower expression (3.8 Miller units) using its native SD sequence. On the contrary, the modified promoters PpsbD-13 and PpsbD-14 gave higher expressions (105.7 and 106.4 Miller units) compared to the native promoter PpsbD-12 (94.6 Miller units). Plac presented a similar expression to a modified PrbcL when using a fluorescent protein GFP reporter (Huang et al. 2010). In our construct, however, the Plac only had moderate expression (34.9 Miller) using the rbs from pXT37a, which was lower than the native cyanobacterial promoters. The strains containing the other modified promoters did not display lacZ expression, suggesting their mutations had negative impacts on protein translation (Supplementary Fig. 3).
In summary, seven strong promoters (PrbcL-12, PpasD-12, PpsaD-12, PatpB-12, PpsbD-12, PpsbD-13 and PpsbD-14), two moderate promoters (PpetE, Plac) and two weak promoters (PpsbA1-12, PpsbA1-13) were screened as molecular tools for metabolic engineering of cyanobacteria. The strong promoters are very helpful for production of non-toxic molecules such like native nutrients and specific biofuels in cyanobacteria. PpetE is a copper inducible promoter and can be used for inducible expression of proteins such as protein degradation tags, etc. The weak promoters and the Plac constructed here enable us to produce toxic products in a limited rate in cyanobacteria.
Production of fatty alcohols in Synechocystis utilizing the constructed platform
Supplementary Table 2 shows the details of the strains and plasmids for fatty alcohol production. Fatty alcohol is one type of promising high-energy molecules to replace the conventional fossil fuels. The biosynthesis of fatty alcohols through heterologously expressing FAR and the effect of environmental stresses on the production of fatty alcohols were investigated in genetically engineered Synechocystis (Tan et al. 2011). However, the yield of fatty alcohols only reached a low level (9.7 ± 2.7 μg/l in shake-flask cultivation) and there is a huge potential to enhance the metabolic flux towards fatty alcohol production by using the promoters and the expression platforms constructed above. Herein, the strong promoter PpsbD-13 and PrbcL-12 were applied for expression of FAR to optimize the fatty alcohol synthesis in cyanobacteria. Meanwhile, we adjusted the enzyme expression levels by co-expression of multi-copies of FAR genes (A. thaliana at3g11980 and the far_jojoba), truncating the peptide sequence of the FAR gene, along with gene knock-out of the agp and phaAB, respectively. Integrating all these genetic modifications based on the molecular tools constructed in this study, the photosynthetic production of fatty alcohols in Synechocystis was dramatically improved.
Synechocystis accumulated more fatty alcohols and alka(e)nes with the utilization of strong promoter Prbc compared to the moderate promoter PpetE (Tan et al. 2011), implying that stronger promoters were preferred to increase the fatty alcohol yield. Besides, expression of multi-copies of genes significantly enhanced protein expression and the product yields in genetically-engineered cyanobacteria. Similarly, expression of multiple copies of ethylene-forming enzyme (efe) doubled ethylene output (Ungerer et al. 2012). Expression of single copy of A. thaliana at3g11980 under PpetE promoter produced approx. 5.3 ± 1.2 μg fatty alcohols/l OD730 in strain Syn-XT34 (Tan et al. 2011); this equals 26.2 ± 5.9 μg/g cell dry weight (CDW). In Fig. 3, the Synechocystis strain Syn-LY10 accumulated 117.5 ± 52.5 μg fatty alcohols/g CDW by expression one copy of PpetE-at3g11980 and one copy of PpetE-jojoba. By contrast, Syn-LY43 containing at3g11980-trunc achieved 426 ± 49.5 μg fatty alcohols/g CDW under regulation of the strong promoter PpsbD13, indicating the peptide truncation greatly improved the enzyme activity of the FAR.

Fatty alcohol productivity in genetically-engineered Synechocystis strains. The detailed strains information is summarized in Supplementary Table 2. The results graphed are averages derived from three independent experiments. The mean values of the fatty alcohol productivity were plotted in the figure (error bars represent the standard deviation)
Glycogen and poly-β-hydroxybutyrate (PHB) synthesis are competing pathways that drive the main metabolic flux away from fatty alcohol synthesis in Synechocystis. Fatty acid production in Synechocystis was markedly improved by cutting off the downstream pathway and knocking-out the competing pathway of PHB (Liu and Sheng 2011). Similarly, the expression of multiple copies of pyruvate decarboxylase gene (pdc) and endogenous alcohol dehydrogenase gene (adh), along with disruption of phaAB gene in PHB pathway, have greatly increased the ethanol productivity by 4.9-fold (Gao et al. 2012), indicating that removing the competing pathway enhanced the metabolic flux towards target products. Herein, we integrated multiple copies of far into the genome and disrupted the phaAB and agp genes. This strategy enhanced fatty alcohol production (Fig. 3). For example Syn-LY25, the strain with one copy of PrbcL12-far_jojoba and disruption of phaAB, generated 569 ± 172 μg fatty alcohols/g CDW. Syn-LY66 yielded 645 ± 108 μg fatty alcohols/g CDW by expression of one copy of Prbc-far_jojoba at the slr0168 locus and one copy of PrbcL12- far_jojoba at the phaAB locus. Strains Syn-LY21 and Syn-XT14C (Supplementary Fig. 2) only obtained a partial disruption of agp; nevertheless, the flux towards fatty alcohols was enhanced. Syn-LY21 generated 50 ± 13.5 μg fatty alcohols/g CDW with expression of PpsaD13-at3g11980 at the agp locus, which was a little higher than that in Syn-XT34 (Tan et al. 2011). Similarly, a greater yield of total fatty alcohols (761 ± 216 μg/g CDW), the highest ever reported up to date, was observed under shake-flask cultivation conditions in Syn-XT14C that contains two copies of far_jojoba and one copy of at3g11980 as well as elimination of the agp and phaAB gene.
Conclusion
An integrative gene expression platform for directional assembly of synthetic parts was successfully developed in the cyanobacterium, Synechocystis PCC 6803. A set of promoter parts were characterized as synthetic tools for metabolic engineering and biotechnological applications in cyanobacteria. Applying these molecular tools, the metabolic flux towards fatty alcohols in Synechocystis was markedly enhanced under flask cultivation condition, and the yield of 761 μg fatty alcohols/g CDW was achieved. This is the first report to modify the fatty alcohol biosynthetic pathway through employing synthetic devices to improve the production of fatty alcohols in cyanobacteria.
References
Boyle PM, Silver PA (2012) Parts plus pipes: synthetic biology approaches to metabolic engineering. Metab Eng 14(3):223–232
Ducat DC, Way JC, Silver PA (2011) Engineering cyanobacteria to generate high-value products. Trends Biotechnol 29(2):95–103
Elhai J, Wolk CP (1988) A versatile class of positive-selection vectors based on the nonviability of palindrome containing plasmids that allows cloning into long polylinkers. Gene 68(1):119–138
Gao H, Tang Q, Xu X (2007) Construction of copper-induced gene expression platform in Synechocystis sp. PCC 6803. Acta Hydrobiol Sin 31(2):240–244
Gao Z, Zhao H, Li Z, Tan X, Lu X (2012) Photosynthetic production of ethanol from carbon dioxide in genetically engineered cyanobacteria. Energy Environ Sci 5(12):9857–9865
Huang HH, Camsund D, Lindblad P, Heidorn T (2010) Design and characterization of molecular tools for a synthetic biology approach towards developing cyanobacterial biotechnology. Nucleic Acids Res 38(8):2577–2593
Liu X, Sheng J, Curtiss III R (2011) Fatty acid production in genetically modified cyanobacteria. Proc Natl Acad Sci USA 108(17):6899–6904
Matteucci M, Heyneker H (1983) Targeted random mutagenesis: the use of ambiguously synthesized oligonucleotides to mutagenize sequences immediately 5′ of an ATG initiation codon. Nucleic Acids Res 11(10):3113–3121
Schaefer MR, Golden SS (1989) Differential expression of members of a cyanobacterial psbA gene family in response to light. J Bacteriol 171(7):3973–3981
Stanier RY, Kunisawa R, Mandel M, Cohen-Bazire G (1971) Purification and properties of unicellular blue-green algae (order Chroococcales). Bacteriol Rev 35(2):171–205
Takeshima Y, Takatsugu N, Sugiura M, Hagiwara H (1994) High-level expression of human superoxide dismutase in the cyanobacterium Anacystis nidulans 6301. Proc Natl Acad Sci USA 91(21):9685–9689
Tan X, Yao L, Gao Q, Wang W, Qi F, Lu X (2011) Photosynthesis driven conversion of carbon dioxide to fatty alcohols and hydrocarbons in cyanobacteria. Metab Eng 13(2):169–176
Tan X, Liang F, Cai K, Lu X (2013) Application of the FLP/FRT recombination system in cyanobacteria for construction of markerless mutants. Appl Microbiol Biotechnol. doi:10.1007/s00253-013-4837-6
Ungerer J, Tao L, Davis M, Ghirardi M, Maness P-C, Yu J (2012) Sustained photosynthetic conversion of CO2 to ethylene in recombinant cyanobacterium Synechocystis 6803. Energy Environ Sci 5(10):8998
Williams JGK (1988) Construction of specific mutations in photosystem II photosynthetic reaction center by genetic engineering methods in Synechocystis 6803. Methods Enzymol 167:766–778
Acknowledgments
We are grateful to the National Basic Research Program of China (973: 2011CBA00907), Knowledge Innovation Program of the Chinese Academy of Sciences (KSCX2-EW-G-1-4 and the Institution-level) and the “100-Talent Program of the Chinese Academy of Sciences” foundation (Grant O91001110A) for the financial support. We appreciate Professor Xudong Xu from Institute of Hydrobiology, Chinese Academy of Sciences for kindly offering the plasmid pHB1567 and pHB1536.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Qi, F., Yao, L., Tan, X. et al. Construction, characterization and application of molecular tools for metabolic engineering of Synechocystis sp.. Biotechnol Lett 35, 1655–1661 (2013). https://doi.org/10.1007/s10529-013-1252-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10529-013-1252-0