
Abstract
The clinical potential of mesenchymal stem cells (MSC) in tissue engineering and regenerative medicine is due to their self-renewal, proliferation and multi-lineage differentiation potential. Clinical use requires large cell numbers; which can, theoretically, be generated by ex vivo expansion of plastic adherent, MSC subpopulation, of bone marrow cells (BMC). Effects of serial culture on MSC phenotype were investigated using non-gel based quantitative proteomic methodology for static monolayer cultures of rat BMC. In total, 382 proteins were relatively quantified (≥2 peptides). Nine proteins were up-regulated and seven down-regulated at passage 4 relative to passage 2 (p ≤ 0.05). We propose that serial culture impacts on MSC expansion (observed decline in colony forming potential and colony size) is through a combination of osteogenic differentiation and ageing/senescence and propose six novel protein biomarkers as candidates for quality control purposes in bioprocessing.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Mesenchymal stem cells (MSC) are the focus of much research interest due to their potential therapeutic use, based on their multi-lineage differentiation potential (they can give rise to myocytes, chondrocytes, adipocytes, and osteogenic cells), and their self-renewal and proliferation capacities, which allow easy ex vivo expansion (Pittenger et al. 1999).
Proteomic techniques provide methods to identify the characteristic protein profiles of MSC populations (Dominici et al. 2006). Thus far, much of MSC proteomic research has used two-dimensional gel electrophoresis (2DE) to quantify and identify changes in the MSCs proteome occurring during serial culture (Sun et al. 2006; Celebi and Elcin 2009), during differentiation, in particular along osteogenic and adipogenic lineages (Lee et al. 2006; Zhang et al. 2007) and to analyse a range of stem cell, MSC and differentiation markers (Park et al. 2007). Although 2DE has yielded useful data, this technique has major limitations since not all proteins can be visualised in this way, particularly large and/or hydrophobic proteins (Maurer and Kuschinsky 2007).
An alternative non-gel-based quantitative MS approach to 2DE is the technique of an isobaric tag for relative and absolute quantitation (iTRAQ). iTRAQ allows simultaneous protein identification and relative quantification in a multiplex format to provide more accurate relative quantification and improved sequence coverage of proteins compared to 2DE (Zhang et al. 2005). All proteins analysed in an iTRAQ study are identified rather a specific subset that is typically analysed by 2DE. An overview of iTRAQ as a proteomics tool and its scope is available elsewhere (Noirel et al. 2011).
This study investigates the impact of serial culture on the expansion of MSC in static cultures systems by analysing the change in protein expression profiles using a high throughput iTRAQ-based proteomic workflow.
Materials and methods
Isolation of rat bone marrow cells (BMC)
Rat BMCs were used rather than human cells due to their reproducibility, ready availability and minimal risk of infection. The BMCs were obtained from the femurs and tibias of Wistar Rats (ages 2–4 months) by flushing out the bone marrow under sterile conditions as previously described (Scutt et al. 2003).
Mesenchymal stem cells (MSC) static monolayer culture
Ten milliliters freshly harvest BMCs were suspended in 10 ml culture medium [DMEM 10 % v/v foetal calf serum (FCS), 2 mM Glutamax and 2 mM penicillin/streptomycin] and seeded into a T75 flask, at 106 cells/ml. The cells were incubated at 37 °C in 7.5 % CO2 and incubated for 4 passages. The first medium change was after 5 days and thereafter twice weekly until ~90 % confluent. Cells were detached using 0.25 % trypsin/1 mM EDTA solution. The detached cells were split 1:2 into new sterile T75-flasks. At passage 2 and 4, cells from 3 confluent flasks were collected for proteomic analysis.
Fibroblastic colony forming unit (CFU-f) and osteogenic differentiation assays
All CFU-f cultures and osteogenic differentiation assays were conducted in triplicate with either 0.3 ml freshly harvested BMCs (passage number 0) or 4 × 103 MSCs (obtained from monolayer cultures after trypsinising) as described by Scutt et al. (2003).
The proteomic workflow using iTRAQ can be found in Fig. 1, and the specific steps are outlined below.

Schematic iTRAQ workflow showing protein extraction, trypsin digest, iTRAQ labelling, peptide fractionation and mass spectrometry
Protein extraction
Pooled and pelleted cells from passage 2 and 4 were re-suspended in 300 μl PBS prior to disruption by probe sonication using the Sonifier 450 (Branson). Soluble proteins were recovered by centrifugation at 21,000×g for 30 min at 4 °C and protein assayed (Glen et al. 2008). Two aliquots of 100 μg of protein from both conditions (cells harvested at passage 2 and 4) were acetone precipitated in preparation for iTRAQ labelling.
Protein digestion and iTRAQ labelling
Peptides were generated by proteolytic digestion using trypsin, following protein reduction, cysteine blocking. These steps and iTRAQ labelling were carried out using the iTRAQ 4-plex kit (Applied Biosystems–now ABSciex) and performed following the manufacturer’s instructions. The two replicate samples from passage 2 were labelled with reagents 114 and 115, and the samples from passage 4 were labelled with the reagents 116 and 117. Afterwards, the labelled samples were combined and vacuum concentrated, fractionated using strong cation exchange and analysed by LC–MS/MS using a QSTAR XL Hybrid ESI Quadrupole time of flight tandem mass spectrometer (Applied Biosystems, Framingham, MA; MDS-Sciex, Concord, Ontario, Canada) coupled with an online capillary liquid chromatography system (Famos, Swichos and ultimate from Dionex/LC Packings, Amsterdam, The Netherlands) as described by Glen et al. (2008).
Protein identification and relative quantification
Protein Pilot software version 2.0 (Applied Biosystems, MDS-Sciex) was used for protein quantification and identification as reported previously (Glen et al. 2008). Identifications were made using rat NCBInr protein database (36,496 entries, downloaded June 2008). Only proteins identified with at least 95 % confidence (ProtScore of 1.3) were reported. Peptides with ≥80 % confidence were used for protein identification. A concatenated target-decoy database search strategy was also employed to estimate the rate of false positives (Boehm et al. 2007), which was calculated to be within the 5 % recommended for reporting proteomic data. Quantification data were parsed into Mathematica version 7.0 (Wolfram, UK). Protein quantifications were obtained by computing the geometric means of the reporters’ intensities. Data was isotope corrected, and bias normalization performed correcting the median ratio of each comparison toward unity.
Functional annotation of proteins
The Panther (Protein analysis through evolutionary relationships) classification system was used to assign molecular function and biological process of proteins. The functional relationship of differentially expressed proteins was further investigated using MetaCore™ (GeneGo Inc.) software.
Statistical analysis
Data for cell cultures and CFU-f assays are expressed as mean values ± standard deviation. Each culture and assay was performed in triplicate. Statistical significance was determined by Student’s t test, with <0.05 considered significant. Changes in protein expression between passage 2 and 4 were qualified using a t test algorithm developed in house (Pham et al. 2010).
Results
Morphology and proliferation rate of bone marrow derived MSC
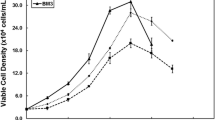
MSCs were expanded in static monolayer cultures by serial passage. Viable cell number, CFU-f and morphology were assessed over 4 passages. From an initial seeding of 4 × 103 adherent cells for Passage 0, the total viable cell number increased continually to 6 × 107 after 4 passages, which represents a 15,000-fold expansion. The cell viability was >99 % for the whole culture period. The time to confluence was reduced for passages 1 to passage 4 inclusive, compared to passage 0, consistent with the finding of Celebi and Elcin (2009) when also analysing rat bone marrow derived MSC. The proliferation rate of MSCs, measured in doubling time, decreased after passage 1 but did not change significantly between passage 2 and 4 (Fig. 2).

Proliferation rate (doubling time) and colony development (CFU-f) for rat bone marrow derived MSCs with increasing passage number. Cells were expanded in static cultures with standard DMEM. Plating density was ~106 cells per T-75 flask. Data are presented as mean ± SD from three different samples
Colony forming potential after each passage is also shown in Fig. 2. An initial increase in colony forming potential observed for passage 1 relative to passage 0 (reflecting enrichment for MSC by adherence to the plastic tissue culture flask following plating out freshly isolated cells) was followed by a decrease in CFU-f over successive passages.
The average colony size also decreased after serial sub-culturing. MSCs expanded in static cultures for 2 passages formed small but recognisable colonies during the CFU-f assay. The colonies formed by MSCs passaged more than twice were barely visible on the plates (Fig. 3). Interestingly, the morphology of the MSC in the monolayer cultures changed over time as the cells became wider and larger (Fig. 3). Osteogenic differentiation potential of passaged MSCs was confirmed by assaying for the presence of alkaline phosphatase, calcium and collagen (Fig. 3), which are biochemical markers of osteoblast differentiation (Scutt et al. 2003).

Results from the methylene blue and osteogenic differentiation assays Methylene blue-stained adherent rat bone marrow cells in the primary culture at a passage level 0, b passage level 2, c passage level 3 and d passage level 4. The four pictures were taken with ~100× magnification. Methylene blue-stained MSCs as CFU-f cultures with colony formation at e passage level 2 and f passage level 4. CFU-f cultures used for osteogenic differentiation assay were stained with g naphtholphosphate and Fast red for alkaline phosphatase, h Alizarin red for calcium and i Sirius red for collagen (I)
Proteomic changes of altered MSC phenotype during culture
The proteomic changes underlying the observed changes in MSC phenotype during serial culture in static culture systems were investigated using an iTRAQ-based workflow. From the iTRAQ data, 23,231 mass spectra were acquired which were identified as 7,134 distinct peptide sequences. A total of 382 proteins were identified by ≥2 peptides. The full peptide and protein lists are shown in Supplementary Table 1. In order to calculate the relative expression levels of the proteins at passage 2 and 4, the mean log10 ratio of the peptides labelled from both replicates at passage 2 (114:114, 115:114) were compared to the mean of the log10 ratios of the peptides labelled from passage 4 (116:114) and (117:114). A total of 16 proteins were differentially expressed with 9 proteins up-regulated and 7 down-regulated (p value 0.05, Bonferroni multiple test correction applied). The lists of the up-regulated proteins and down-regulated protein are shown in Table 1 with their functional annotation (Huang et al. 2008, 2009).
Discussion
The effect of serial subculturing from static cultures on the proteome of rat bone marrow derived MSCs has been determined: whilst the overall proliferation rate of MSCs only decreased slightly over 4 passages (Fig. 2), the effects on CFU-f potential (Fig. 2) and the morphology of the MSC cultures (Fig. 3) were more marked. After 4 passages, a change in the cells morphology can be observed as they became wider, which together with decreased colony forming ability, can indicate ageing of the culture (Stolzing and Scutt 2006).
The proteome of MSCs from passage 2 was compared to that of MSCs from passage 4, to determine alterations in protein expression associated with serial culturing and loss of expansion potential. Cells from passage 2 were chosen in preference to passage 1 in order to allow the MSC to adapt to in vitro conditions and adopt a stable phenotype. Previous reports had suggested that differentiation and proliferation capacity remained static from passage 2 to 10 (Bruder et al. 1997). However, the CFU-f assay data showed a dramatic decrease in colony numbers at passage 4, and because of this, passages 2 and 4 were chosen for comparison.
In general, there is agreement on proteins involved in serial culturing, ageing and osteogenic differentiation (Table 1) in our study compare to other MSC studies (Sun et al. 2006; Celebi and Elcin 2009; Kim et al. 2008). For example, the up-regulation of Annexin A2 at passage 4 relative to passage 2 has also been demonstrated in human bone marrow derived MSC during serial culturing. The glycolytic enzymes enolase and pyruvate kinase were also shown to be up-regulated in human BMC derived MSC (Sun et al. 2006). Whilst this current study did not identify these particular enzymes, a related enzyme on the same metabolic pathway, triose phosphate isomerase was up-regulated (Table 1).
The up-regulation of the proteins nuclear transport factor 2 and karyopherin (also known as importin) at passage 4 compared to passage 2 represent novel findings–these are components of the RAN transport pathway (Table 1). In terms of functional significance, the RAN transport pathway acts as a molecular switch for the karyopherin to regulate nuclear transport during cell cycle, interphase cells (Yudin and Fainzilber 2009). Another 4 other proteins that were down-regulated: the cysteine and glycine-rich protein 1, AHNAK nucleoprotein, ribosomal protein S27and glutamyl-prolyl-tRNA synthetase are also novel protein changes associated with serial culture (Table 1).
The list of proteins that were up-regulated and down-regulated between passages 2 and 4 were further analysed using the Metacore platform (GeneGo Inc., St. Joseph, MI). Metacore provides an interface for submission of proteins in terms of gene identity for searching against an extensive, manually curated database of proteins involved in biological networks associated with transcriptional and metabolic regulation, and in cell signalling incorporating published protein–protein, protein–DNA interactions (www.genego.com). The network analysis identified 7 networks (regulatory and metabolic), of which the 3 most highly significant (5 or more root nodes, Z score of >40) involved the protein components of
1. Ubiquitin (Rps27a), alpha actinin 4, nuclear transport factor 2, karyopherin alpha 1,
2. Ubiquitin (Rps27a), IQGAP1, karyopherin alpha 1, AHNAK,
3. Annexin A2, cyclophilin A, AHNAK and calponin 1.
The top 3 regulatory networks were RAN transport (as mentioned above) cell cycle and glycolysis. Of note, the glycolytic enzymes, triose phosphate isomerase and phosphoglycerate mutase were found to be altered in this study, suggesting alterations in energy metabolism, consistent with other studies (Sun et al. 2006).
In summary, the utility of the iTRAQ workflow has been demonstrated as a useful tool to investigate the change in phenotype of MSC during serial culture, and has generated results that are both novel and complementary to previous 2DE proteomic studies on MSCs. Crucially, the data highlights the impact of the culture condition of the MSC with significant loss of self-renewal capacity with repeated serial subculture. Taking all these findings together, the observed reduction in CFU-f in this study could be explained by a combination of osteogenic differentiation and with signs of potential ageing of cells in the culture.
The use of iTRAQ to identify key up-and down-regulated proteins related to the loss of self-renewal capacity during culture expansion, could contribute to the development of a panel of biomarkers for monitoring cell status. Such a panel could in turn be used as a monitor of quality control, assessing the validity of the quality of large scale MSC preparations intended for tissue engineering purposes.
References
Boehm A, Putz S, Altenhofer D, Sickmann A, Falk M (2007) Precise protein quantification based on peptide quantification using iTRAQTM. BMC Bioinformatics 8:214–232
Bruder SP, Jaiswal N, Haynesworth SE (1997) Growth kinetics, self-renewal, and the osteogenic potential of purified human mesenchymal stem cells during extensive sub-cultivation and following cryopreservation. J Cell Biochem 64:278–294
Celebi BL, Elcin YM (2009) Proteome analysis of rat bone marrow mesenchymal stem cell subcultures. J Proteome Res 8:2164–2172
Dominici M, Le Blanc K, Mueller I, Slaper-Cortenbach I, Marini F, Krause D, Deans R, Keating A, Prockop Dj, Horwitz E (2006) Minimal criteria for defining multipotent mesenchymal stromal cells.The international society for cellular therapy position statement. Cytotherapy 8:315–317
Glen A, Gan CS, Hamdy FC, Eaton CL, Cross SS, Catto JW, Wright PC, Rehman I (2008) iTRAQ-facilitated proteomic analysis of human prostate cancer cells identifies proteins associated with progression. J Proteome Res 7:897–907
Huang DW, Sherman BT, Lempicki RA (2008) Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4:44–57
Huang DW, Sherman BT, Lempicki RA (2009) Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acid Res 37:1–13
Kim JS, Lee HK, Kim MR, Kim PK, Kim CW (2008) Differentially expressed proteins of mesenchymal stem cells derived from human umbilical cord blood during osteogenic differentiation. Biosci Biotechnol Biochem 72:2309–2317
Lee HK, Lee BH, Park SA, Kim CW (2006) The proteomic analysis of an adipocyte differentiated from human mesenchymal stem cells using two-dimensional gel electrophoresis. Proteomics 6:1223–1229
Maurer MH, Kuschinsky WK (2007) Proteomics. In: Lajtha A, Gibson GE, Dienel GA (eds) Handbook of neurochemistry and molecular neurobiology, vol 5., brain energetics. Integration of cellular and molecular processesSpringer, New York, pp 737–769
Noirel J, Evans C, Salim M, Mukherjee J, Yen OS, Pandhal J, Pham TK, Biggs CA, Wright PC (2011) Methods in quantitative proteomics: setting iTRAQ on the right track. Curr Proteomics 8:17–30
Park HW, Shin JS, Kim CW (2007) Proteome of mesenchymal stem cells. Proteomics 7:2881–2894
Pham TK, Roy S, Noirel J, Douglas I, Wright PC, Stafford GP (2010) A quantitative proteomic analysis of biofilm adaptation by the periodontal pathogen Tannerella forsythia. Proteomics 10:3130–3141
Pittenger MF, Mackay AM, Beck SC, Jaiswal RK, Douglas R, Mosca JD, Moorman MA, Simonetti DW, Craig S, Marshak DR (1999) Multilineage potential of adult human mesenchymal stem cells. Science 284:143–147
Scutt A, Reading L, Scutt N, Still K (2003) Mineralizing fibroblast-colony-forming assays. Methods Mol Med 80:29–39
Stolzing A, Scutt A (2006) Age-related impairment of mesenchymal progenitor cell function. Ageing Cell 5:213–224
Sun HJ, Bahk YY, Choi YR, Shim JH, Han SH, Lee JW (2006) A proteomic analysis during serial subculture and osteogenic differentiation of human mesenchymal stem cell. J Orthop Res 24:2059–2071
Yudin D, Fainzilber M (2009) RAN on tracks––cytoplasmic roles for a nuclear regulator. J Cell Sci 122:587–593
Zhang Y, Wolf-Yadlin A, Ross PL, Pappin DJ, Rush J, Lauffenburger DA, White FM et al (2005) Time-resolved mass spectrometry of tyrosine phosphorylation sites in the epidermal growth factor receptor signaling network reveals dynamic modules. Mol Cell Proteomics 4:1240–1250
Zhang AX, Yu WH, Ma BF, Yu XB, Mao FF, Liu W, Zhang JQ, Zhang XM, Li SN, Li MT, Lahn BT, Xiang AP (2007) Proteomic identification of differently expressed proteins responsible for osteoblast differentiation from human mesenchymal stem cells. Mol Cell Biochem 304:167–179
Acknowledgments
The authors wish to acknowledge the UK Engineering and Physical Sciences Research Council (EPSRC) for an Advanced Research Fellowship for Biggs (EP/E053556/01) and further funding for the ChELSI Institute (EP/E036252/1). The authors also wish to acknowledge The University of Sheffield for a studentship for Stefan Peter.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Peter, S., Evans, C., Ow, S.Y. et al. Proteomic analysis of the impact of static culturing on the expansion of rat bone marrow mesenchymal stem cells. Biotechnol Lett 34, 1589–1596 (2012). https://doi.org/10.1007/s10529-012-0935-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10529-012-0935-2