
Abstract
In the present study, an endochitinase gene, Lbchi32, was cloned from Limonium bicolor. The cDNA sequence of Lbchi32 was 1,443 bp in length and encoded 319 amino acid residues. The DNA sequence of Lbchi32 was 2,512 bp in length and contained three exons and two introns. The Lbchi32 gene was inserted into a pPIC9 vector and transferred into Pichia pastoris strains GS115 and KM71 for heterologous expression. SDS-PAGE analyses indicated that LbCHI32 was expressed in both GS115 and KM71 and that it was secreted extracellularly. The optimal reaction conditions for LbCHI32 activity are 45°C, pH 5.0, and 5 mM Ba2+. The LbCHI32 enzyme can efficiently degrade chitin, chitin derivatives, and the cell walls of different pathogenic fungi, including phytopathogenic Rhizoctonia solani, Fusarium oxysporum, Sclerotinia sclerotiorum, Valsa sordida, Septoria tritici, and Phytophthora sojae. These findings suggest that Lbchi32 has potential use in the degradation of chitin and chitin derivatives.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Chitin, which is a polymer of N-acetylglucosamine, is the second most abundant natural polymer on earth after cellulose. It is currently attracting considerable attention as an unutilized biomass resource. Chitinase can hydrolyze chitin to chitin oligosaccharides, including oligomers, chitobiose, and N-acetylglucosamine, which can be utilized as carbon or nitrogen sources (Schrempf 2001). This suggests that chitinase can play an important role in the utilization of chitin. In addition, chitin is also the main component of the cell walls of phytopathogenic fungi. Chitinase can catalyze the hydrolysis of fungal cell walls and thereby exhibits antifungal activities. Accordingly, this enzyme could be used directly as an antifungal agent against fungal phytopathogens (Robert et al. 2002; Ohnuma et al. 2004). Therefore, there is the potential for chitinases to play important roles in both industrial production and the biocontrol of plant diseases.
Chitinases are widely distributed in various organisms; among these, plant chitinases comprise a large family. Previous studies have shown that chitinases are involved in plant defense mechanisms against fungal pathogens. For instance, the chitinase (Chi1) from Triticum aestivum “Ning7840” has been found to be involved in a generalized resistance response to Fusarium graminearum, suggesting an antifungal role (Kong et al. 2005). A chitinase gene from mung bean (Phaseolus mungo) can exert antifungal activity against various fungi, including Fusarium solani, F. oxysporum, Mycosphaerella arachidicola, Pythium aphanidermatum, and Sclerotium rolfsii (Ye and Ng 2005). Overexpression of Mcchit1, a chitinase gene isolated from Momordica charantia, in Nicotiana benthamiana was found to improve resistance significantly to the plant pathogenic fungus Phytophthora nicotianae. Furthermore, Mcchit1-transformed cotton exhibits elevated resistance to Verticillium wilt, indicating that the Mcchit1 gene is a useful gene in the engineering of plant resistance to fungal diseases (Xiao et al. 2007). Also, a chitinase gene (Vvchit1a) from Vitis vinifera was induced in V. vinifera leaves infected with Bacillus cinerea or Pseudomonas syringae pv. pisi, a bacterium that induces an incompatible interaction (Robert et al. 2002).
Determination of the chitin and chitin derivative degradation activity is very important in characterizing chitinases. Previous studies of plant chitinases, however, have focused mainly on their roles as antifungal agents, and little research had been performed on plant chitinase activities with respect to the degradation of chitin and chitin derivatives.
In the present study, we cloned a chitinase gene, Lbchi32, from Limonium bicolor, a biennial flowering plant belonging to the Plumbaginaceae. To investigate the role of Lbchi32 in decomposing chitin, chitin derivatives, and the fungal cell walls, we expressed the gene in the yeast Pichia pastoris. We found that the recombinant LbCHI32 protein produced by transgenic yeast can exhibit high levels of activity in the degradation of chitin resources and fungal/oomycete cell walls. Our results suggest that plant chitinases, similarly to fungal chitinase, also exhibit high decomposing activity against chitin and chitin derivatives and thus have potential value in chitin exploitation. Further, the ability of LbCHI32 to decompose the cell walls of different fungi suggests that the Lbchi32 gene is also involved in antifungal responses in L. bicolor.
Materials and Methods
Plant Material, Strains and Culture Conditions
Limonium bicolor seedlings were grown in pots containing a mixture of turf peat and sand (2:1 v/v). Plants were grown in a greenhouse under controlled conditions of 65–75% relative humidity, 14 h light, and an average temperature of 24°C. The leaves of L. bicolor were harvested for DNA and RNA extractions.
Two P. pastoris strains, GS115 (Mut+, His−) and KM71(MutS, His−) (Invitrogen, Carlsbad, USA), were used as the hosts for expression of the Lbchi32 chitinase gene. Five phytopathogenic fungal strains, including F. oxysporum (soybean root rot), Septoria tritici (wheat leaf wither), Valsa sordida (poplar bark rot), Rhizoctonia solani (rice sheath blight), Sclerotinia sclerotiorum (sclerotium disease on soybean), and one strain of oomycete plant pathogen, Phytophthora sojae (soybean root rot), were obtained from the Microbial Genetic Engineering Laboratory, Harbin Institute of Technology, Harbin, China. The mycelia of the five phytopathogenic fungal strains were inoculated into potato dextrose medium and incubated at 25°C for 48 h with shaking at 180 rpm. Mycelium of the oomycete strain was inoculated into carrot medium and incubated at 25°C for 48 h with shaking at 180 rpm. After cultivation, the mycelia of these strains were harvested. Cell wall fragments were prepared from the mycelia of each strain, according to the method of Morissette et al. (2006), and were used as substrates for fungal cell wall degradation analyses.
Cloning of the Chitinase Lbchi32 Gene
A cDNA library was constructed from the leaves of L. bicolor, and 2358 ESTs were obtained by random sequencing of the library clones (Wang et al. 2008). The EST (GenBank no. EH793640) containing a full-length open reading frame (ORF) of endochitinase Lbchi32 was identified from the cDNA library by EST analysis. Primers for amplification of the DNA sequence of Lbchi32 were designed as follows: chi32-1, 5′-ATGGGGAGGCATTGGAGACTGGT-3′; chi32-2, 5′-TTATGACGATGCAGAGCCGGATG-3′. Total DNA was isolated from L. bicolor leaves according to the procedure described by Sambrook et al. (2002), and was used as a PCR template. The PCR product was cloned into a pMD18-T vector (Takara, Dalian, China) for sequencing.
Bioinformatics Analysis of the Lbchi32 Gene
The catalytic domain of the LbCHI32 protein was identified by the BlastP program of NCBI, and the theoretical molecular mass and isoelectric point of the LbCHI32 protein were calculated using the ProtParam tool (http://us.expasy.org/tools/protparam.html). A signal peptide was predicted using SignalP3.0 Server (http://www.cbs.dtu.dk/services/SignalP/) and glycosylation sites were predicted using the ELM Server (http://elm.eu.org/). Amino acid sequences of chitinases from various plant species (including L. bicolor) were aligned by the ClustalX program.
Construction of Recombinant pPIC9-chi32 and Yeast Transformation
The primers for amplification of the ORF of the Lbchi32 gene were designed as follows: chi32-L, 5′-ATCGCTCGAGAAAAGAGAGGCTGAAGCTTACGCGCTGGACCTGACGGACCAGCTCG-3′ (XhoI site underlined); chi32-R, 5′-CGATGCGGCCGCTGACGATGCAGAGCCGGATGGGTTG-3′ (NotI site underlined). The ORF of the Lbchi32 gene was cloned into a pPIC9 expression vector (Invitrogen, Carlsbad, USA), and the recombinant vector (pPIC9-chi32) was transformed into GS115 or KM71 cells by pulsed electroporation using a Bio-Rad GenePulser (1150 V, 25IF, 200×).
SDS-PAGE Analysis of Recombinant Chitinase LbCHI32
Three types of yeast transformants, the GS115 strain harboring pPIC9-chi32 (GS115-chi32), the KM71 strain harboring pPIC9-chi32 (KM71-chi32), and yeast transformants not harboring pPIC9 (GS115-pPIC9, used as a control), were cultured following the procedures described in the Pichia expression kit manual (K1710-01; Invitrogen). Methanol was added to the culture medium at a final concentration of 1% (v/v) every 24 h to maintain the induction. The supernatants were harvested after culturing for 1, 2, 3, 4, 5, and 6 days at 30°C. After the addition of 1× loading buffer, the supernatants were boiled for 5 min, centrifuged for 10 min at 8,000 rpm, and loaded onto a 12% SDS-PAGE gel.
Assay of LbCHI32 Activity
Chitinase activity was measured according to Liu et al. (2008). The strain transformed with empty pPIC9 was induced by 1% (v/v) methanol at 30°C for 1–7 days at 1-day intervals, and the fermentation medium was harvested for measurement of chitinase activity.
The yeast transformant GS115-chi32 was induced by 1% (v/v) methanol at 30°C for 6 days. The yeast cells were centrifuged, and the supernatant was used in the following experiments. For determination of the optimum temperature for LbCHI32 activity, the reaction mixtures (pH 4.5) were incubated at 35–80°C for 30 min at 5°C intervals, boiled for 5 min, supplemented with 0.05% (w/v) potassium ferrocyanide, and boiled for 10 min. The supernatant (enzyme solution) was incubated at 35–80°C for 30 min at 5°C intervals (pH 4.5) and chilled on ice; the remaining activity was then measured. Determination of the optimum pH for LbCHI32 enzyme activity was performed at 50°C over a pH range of 3–12, at intervals of 1 pH unit. The pH of the reaction mixture was adjusted using a series of buffers: 0.01 M citric acid–0.02 M Na2HPO4 (pH 3–5), 0.02 M Na2HPO4–NaH2PO4 (pH 6–8), 0.02 M glycine–NaOH (pH 9–10), and 0.05 M Na2HPO4–0.01 M NaOH (pH 11–12).
The Effect of Ions on LbCHI32 Activity
LbCHI32 activity was measured in a reaction buffer that was supplemented with 5 mM of a metal ion in the form of a chloride or sulfate salt. Different buffer solutions were prepared, each spiked with a different metal salt (KCl, BaCl2, CaC12, CoCl2, NaCl, LiCl, MgSO4, MnSO4, FeSO4, CuSO4, or ZnSO4). The relative activity of LbCHI32 was calculated as the activity in the ion-enhanced buffer divided by the activity of LbCHI32 under normal conditions (the control).
Chitin Derivative and Fungal Cell Wall Degradation Analyses
The substrates used for LbCHI32 degradation analysis, including 1% cell wall fragments from each fungus and 1% of chitin derivatives (chitosan, N.O-Carb-Chitosan, colloidal chitin, or N.O-Carb-chitin), were dissolved in 0.02 M HAc-NaAc buffer (pH 4.5). Cell wall fragments from the six fungal/oomycete plant pathogens (R. solani, F. oxysporum, V. sordida, Sclerotinia sclerotiorum, Septoria tritici, and Phytophthora sojae) were prepared as described by Morissette et al. (2006). The chitinase activity of LbCHI32 toward the substrates was measured using the Schales method (Imoto and Yagishita 1971).
All of the above experiments were completed, at a minimum, in triplicate, and average values were calculated based on results from three independent experiments. Data were analyzed using SPSS version 11.0 statistical software (SPSS Inc., Chicago, IL). Sample variability is given as the standard deviation.
Results
Cloning and Sequence Analysis of Chitinase Lbchi32
The cDNA sequence of Lbchi32 is 1,443 bp in length, and the ORF length is 960 bp. It encodes 319 amino acids with a calculated molecular mass of 32.4 kDa and a predicted isoelectric point of 5.38. The DNA sequence of the Lbchi32 chitinase gene was 2,512 bp in length and contained three exons and two introns. The lengths of the three exons were 391, 842, and 379 bp, and the two introns were 1,203 and 349 bp. The splice sites between exons and introns followed the GT–AG splicing rule. The cDNA and DNA sequences of the Lbchi32 gene have been deposited in the GenBank database under accession numbers DQ431249 and EU744179, respectively.
SignalP prediction showed a signal peptide (25 amino acids) in the N-terminal sequence of LbCHI32. The LbCHI32 protein (ABD92820) is predicted to be cleaved by a signal peptidase between positions A25 and L26 (SAA|||LDL). There are three potential N-linked glycosylation sites, at C57-P62, Q208-A213, and K244-S249 of the LbCHI32 amino acid sequence. Pfam analysis revealed that the LbCHI32 protein had a catalytic domain common to members of the family 19 chitinases (InterPro acc. no.: IPR000726), indicating that it is a member of the family 19 glycosyl hydrolases (endochitinase).
Analysis of Sequence Identity
BlastP prediction showed that the catalytic domain of LbCHI32 consists of approximately 220–230 amino acid residues and three catalytic residues (E128, E150, Y183) (Fig. 1). To determine the conservation of the three catalytic residues, multiple sequence alignment of the chitinases from 21 plant species was conducted using the ClustalX program. This revealed that the amino acid sequences of these chitinases were conserved, with identities ranging from 71 to 80%. Furthermore, the chitin-catalyzing domains of the different chitinases were highly conserved, with the three catalytic residues (E, E, and Y) being particularly highly conserved (Fig. 1).

Catalytic residues of chitinase Ib from 21 plant species. BAH30273, NP_172076, AAL06524: chitinases from Arabidopsis thaliana; ABV89660: chitinase from Brassica rapa; AAP80800, AAQ56598, AAQ56599: chitinases from Gossypium hirsutum; EEF43061, EEF46716: chitinases from Ricinus communis; XP_002316022, ABK95866: chitinases from Populus trichocarpa; ABL86685, ABL86686: chitinases from Gossypium barbadense; ACM45714, NP_188317: chitinases from Pyrus pyrifolia; XP_002276563, XP_002269474: chitinases from Vitis vinifera; ABN08775: chitinase from Medicago truncatula; ABD92820: chitinase from Limonium bicolor; ACN39823: chitinase from Picea sitchensis; EAZ07679: chitinase from Oryza sativa. Three highly conserved catalytic residues (E, E, and Y) are boxed. Asterisk (*), identity. Colon (:), high similarity. Period (.), low similarity
Heterologous Expression of Lbchi32 in Pichia pastoris
Pichia pastoris GS115 and KM71 transformants harboring pPIC9-chi32 were generated, and the expression of Lbchi32 was induced by methanol. After induction culture, SDS-PAGE analysis was conducted to determine whether the LbCHI32 protein was synthesized in P. pastoris. Compared with the control P. pastoris transformed with an empty pPIC9, both the GS115 and KM71 transformants showed a clearly visible protein band with a molecular mass of approximately 32 kDa on an SDS-PAGE gel (Fig. 2). This result indicates that the LbCHI32 protein was successfully synthesized in the GS115 and KM71 yeast cells, and that it was secreted extracellularly.

SDS-PAGE analysis of the heterologous expression of Lbchi32 in the GS115 and KM71 strains of Pichia pastoris. Top panel, Pichia pastoris GS115 transformed with Lbchi32 (GS115-chi32). Bottom panel, P. pastoris KM71 transformed with Lbchi32 (KM71-chi32). Lane 1, supernatant of P. pastoris transformed with empty pPIC9 induced for 1 day. Lane 2, protein marker. Lane 3, supernatant of P. pastoris transformed with empty pPIC9 induced for 4 days. Lanes 4–9, supernatants of the recombinant yeasts (GS115-chi32 or KM71-chi32) after induction for 1, 2, 3, 4, 5, and 6 days, respectively
Properties of Recombinant LbCHI32
Chitinase activity was not detected in the culture medium of P. pastoris GS115 transformed with an empty pPIC9 vector after methanol induction, indicating that the chitinase detected in transgenic yeast cells is due to the expression of the exogenous gene Lbchi32.
Both the GS115 and KM71 transformants harboring Lbchi32 showed chitinase activity; however, the GS115 yeast exhibited a higher chitinase activity than the KM71 yeast (Fig. 3). The chitinase activity of the LbCHI32 transgenic yeast strain GS115-chi32 showed a peak value at 6 days following methanol induction, with a chitinase activity of 0.71 U/ml (Fig. 3). The transformant that expressed the chitinase at the highest level (named GS115-chi32) was selected for further experimentation. LbCHI32 activity reached a peak at 45°C and was dramatically reduced at temperatures over 55°C. When the temperature reached 60°C, all enzyme activity was lost. Therefore, the optimal reaction temperature for LbCHI32 was determined to be 45°C (Fig. 3). The enzyme activity of recombinant LbCHI32 was stable when incubated between 35 and 65°C for 30 min, and decreased rapidly when temperatures exceeded 60°C. The chitinase was inactivated when incubated at 65°C for 30 min (Fig. 3). At 45°C, the enzyme activity of recombinant LbCHI32 was stable at pHs between 3 and 10, with a peak of activity at pH 5. Chitinase activity was reduced rapidly when the pH exceeded 10 and was inactivated when the pH reached 11 (Fig. 3).

Activities and properties of recombinant chitinase LbCHI32. a Chitinase activity of Pichia pastoris GS115-chi32 (diamond) and KM71-chi32 (square) at different induction times. b Effect of temperature on the chitinase activity of LbCHI32. c Effect of temperature on the chitinase stability of LbCHI32. d Effect of pH on the chitinase activity of LbCHI32. Error bars represent the standard deviation for three independent experiments
The activity of recombinant LbCHI32 was strongly inhibited by Mn2+ and K+ but was stimulated by Ba2+ and Cu2+; the activity of LbCHI32 reached 0.86 U/ml when activated by Ba2+ and 0.77 U/ml by Cu2+. Co2+, Na+, Li+, Mg2+, and Zn2+ did not induce any substantive activation of LbCHI32 (Fig. 4).

Effect of metal ions on the chitinase activity of LbCHI32. Error bars represent the standard deviation for three independent experiments
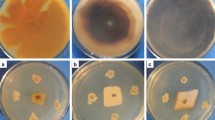
The activity of recombinant LbCHI32 toward different substrates was measured. LbCHI32 showed hydrolytic activity against all the test substrates, indicating that LbCHI32 can decompose the tested substrates. The activity of LbCHI32 toward different substrates ranged from 0.73 to 0.47 U/ml (Fig. 5).

Activity of chitinase LbCHI32 on substrates: 1, N.O-Carb-chitin; 2, Colloidal chitin; 3, Chitosan; 4, N.O-Carb-Chitosan; 5–10, cell wall chitin of Sclerotinia sclerotiorum, Fusarium oxysporum, Rhizoctonia solani, Phytophthora sojae, Septoria tritici, and Valsa sordida. Error bars represent the standard deviation for three independent experiments
The optimal reaction system for LbCHI32 appears to comprise a temperature of 45°C (Fig. 3), a pH of 5.0 (Fig. 3), and a Ba2+ concentration of 5 mM (Fig. 4). Using this reaction system, the highest activity achieved by the recombinant chitinase LbCHI32 was 1.09 U/ml when the cell wall chitin of the fungal pathogen F. oxysporum was used as a substrate.
Discussion
Pfam prediction showed that LbCHI32 belongs to glycoside hydrolase family 19 (Fig. 1). Chitinases of family 19 (also known as classes IA/I and IB/II) are enzymes from plants that function in the defense against fungal and insect pathogens by destroying their chitin-containing cell wall. Class IA/I and IB/II enzymes differ with respect to the presence (IA/I) or absence (IB/II) of an N-terminal chitin-binding domain (Hahn et al. 2000; Fukamizo 2000; Robertus and Monzingo 1999; Robertus et al. 1998; Monzingo et al. 1996). LbCHI32 lacks an N-terminal chitin-binding domain and therefore belongs to class IB.
In eukaryotic cells, N-glycosylation is the most common modification of secretory and membrane-bound proteins. N-linked glycosylation is a cotranslational process involving the transfer of an oligosaccharide chain to an asparagine residue in the protein (Apweiler et al. 1999; Mellquist et al. 1998; Nilsson and von Heijne 2000; Shakin-Eshleman et al. 1996). Kasturi et al. (1997) showed that amino residues Trp, Asp, and Glu are uncommon before the Ser/Thr position. Efficient glycosylation usually occurs when ~60 residues or more separate the glycosylation acceptor site from the C terminus. According to an ELM program prediction, three N-glycosylation sites (C57-P62, Q208-A213, and K244-S249) occur in the LbCHI32 protein, suggesting that glycosylation may occur in these regions. Furthermore, after glycosylation modification, the molecular mass of the LbCHI32 protein is larger than that predicted. Similarly, recombinant BjCHI1 and BjCHI2 in P. pastoris showed obviously larger molecular masses than those calculated from SDS-PAGE, and the larger masses were due to glycosylation of the enzyme (Tang et al. 2004). In the present study, the recombinant LbCHI32 exhibited a molecular mass larger than the 32 kDa observed on SDS-PAGE (Fig. 2). Therefore, this result indicates that LbCHI32 might be modified by glycosylation or some other post-translational modification.
In the present study, to assay the enzyme activity of the recombinant LbCHI32, four types of chitin derivative and six types of phytopathogen cell wall were used as substrates. The results showed that the decomposing ability of the LbCHI32 enzyme toward different chitin derivatives was similar to that toward different phytopathogen cell walls (Fig. 5). Therefore, these data indicate that Lbchi32 not only probably plays a role in fungal plant pathogen defense but also has the potential to decompose organic chitin resources.
The effect of metal ions on chitinase activities has been studied previously. For instance, the activity of a chitinase from Bacillus sp. DAU101 was strongly inhibited by Zn2+, Cu2+, and Hg2+ but stimulated by Co2+ (Lee et al. 2007). Similarly, the activity of two chitinases from Paecilomyces variotii DG-3, chi32 and chi46, can be enhanced by Co2+ (Nguyen et al. 2009). In contrast, the activity of chitinase from Chaetomium cupreum was inhibited by Co2+ and Zn2+ and enhanced by Mg2+, K2+, and Ba2+ (Wang and Yang 2009). The activity of an Enterobacter aerogenes chitinase was activated by Ba2+, Ca2+, Mn2+, and Zn2+ but was highly inhibited by Co2+ and Mg2+ (Tang et al. 2001). In the present study, the activity of LbCHI32 was strongly inhibited by Mn2+ and K+, stimulated by Ba2+ and Cu2+, and not affected by Co2+, Na+, Li+, Mg2+, and Zn2+. These results indicate that the activity of chitinase can be strongly influenced (activated or inhibited) by metal ions; however, the same ion may have different effects on the activity of different chitinases. Therefore, it is important to determine the effects of metal ions on chitinase activity for the characterization of a particular chitinase.
In conclusion, a chitinase gene was cloned from L. bicolor and its chitinase activity was characterized. Expression of the Lbchi32 gene in P. pastoris can provide valuable information on the development of a new, cost-effective technique for large-scale production of LbCHI32. In view of the need for safer and environmentally friendly biocontrol tools, the recombinant chitinase LbCHI32 produced in P. pastoris has the potential to be used agriculturally in the biocontrol of fungal plant pathogens.
References
Apweiler R, Hermjakob H, Sharon N (1999) On the frequency of protein glycosylation, as deduced from analysis of the SWISS-PROT database. Biochim Biophys Acta 1473:4–8
Fukamizo T (2000) Chitinolytic enzymes: catalysis, substrate binding, and their application. Curr Protein Pept Sci 1:105–124
Hahn M, Hennig M, Schlesier B, Höhne W (2000) Structure of jack bean chitinase. Acta Crystallogr D Biol Crystallogr 56:1096–1099
Imoto T, Yagishita K (1971) A simple activity measurement of lysozyme. Agric Biol Chem 35:1154–1156
Kasturi L, Chen HG, Shakin-Eshleman SH (1997) Regulation of N-linked core glycosylation: use of a site-directed mutagenesis approach to identify Asn-Xaa-Ser/Thr sequons that are poor oligosaccharide acceptors. Biochem J 323:415–419
Kong L, Anderson JM, Ohm HW (2005) Induction of wheat defense and stress-related genes in response to Fusarium graminearum. Genome 48:29–40
Lee YS, Park IH, Yoo JS, Chung SY, Lee YC, Cho YS, Ahn SC, Kim CM, Choi YL (2007) Cloning, purification, and characterization of chitinase from Bacillus sp. DAU101. Bioresour Technol 98:2734–2741
Liu ZH, Yang Q, Hu S, Zhang JD, Ma J (2008) Cloning and characterization of a novel chitinase gene (chi46) from Chaetomium globosum and identification of its biological activity. Appl Microbiol Biotechnol 80:241–252
Mellquist JL, Kasturi L, Spitalnik SL, Shakin-Eshleman SH (1998) The amino acid following an asn-X-Ser/Thr sequon is an important determinant of N-linked core glycosylation efficiency. Biochemistry 37:6833–6837
Monzingo AF, Marcotte EM, Hart PJ, Robertus JD (1996) Chitinases, chitosanases, and lysozymes can be divided into procaryotic and eucaryotic families sharing a conserved core. Nat Struct Biol 3:133–140
Morissette DC, Seguin P, Jabaji-Hare SH (2006) Expression regulation of the endochitinase-encoding gene sechi44 from the mycoparasite Stachybotrys elegans. Can J Microbiol 52:1103–1109
Nguyen VN, Oh IJ, Kim YJ, Kim KY, Kim YC, Park RD (2009) Purification and characterization of chitinases from Paecilomyces variotii DG-3 parasitizing on Meloidogyne incognita eggs. J Ind Microbiol Biotechnol 36:195–203
Nilsson I, von Heijne G (2000) Glycosylation efficiency of Asn-Xaa-Thr sequons depends both on the distance from the C terminus and on the presence of a downstream transmembrane segment. J Biol Chem 275:17338–17343
Ohnuma T, Taira T, Yamagami T, Aso Y, Ishiguro M (2004) Molecular cloning, functional expression, and mutagenesis of cDNA encoding class I chitinase from rye (Secale cereale) seeds. Biosci Biotechnol Biochem 68:324–332
Robert N, Roche K, Lebeau Y, Breda C, Boulay M, Esnault R, Buffard D (2002) Expression of grapevine chitinase genes in berries and leaves infected by fungal or bacterial pathogens. Plant Sci 162:389–400
Robertus JD, Monzingo AF (1999) The structure and action of chitinases. EXS 87:125–135
Robertus JD, Monzingo AF, Marcotte EM, Hart PJ (1998) Structural analysis shows five glycohydrolase families diverged from a common ancestor. J Exp Zool 282:127–132
Sambrook J, Frisch EF, Maniatis T (2002) Molecular cloning: a laboratory manual. Cold Spring Harbor Laboratory Press, Long Island, NY
Schrempf H (2001) Recognition and degradation of chitin by streptomycetes. Antonie van Leeuwenhoek 79:285–289
Shakin-Eshleman SH, Spitalnik SL, Kasturi L (1996) The amino acid at the X position of an Asn-X-Ser sequon is an important determinant of N-linked core-glycosylation efficiency. J Biol Chem 271:6363–6366
Tang Y, Zhao J, Ding S, Liu S, Yang Z (2001) Purification and properties of chitinase from Enterobacter aerogenes. Acta Microbiol Sin 41:82
Tang CM, Chye S, Ouyang SW, Zhao KJ, Ubhayasekera W, Mowbray SL (2004) Functional analyses of the chitin-binding domains and the catalytic domain of Brassica juncea chitinase BjCHI1. Plant Mol Biol 56:285–298
Wang YJ, Yang Q (2009) Cloning and expression of a novel chitinase chi58 from Chaetomium cupreum in Pichia pastoris. Biochem Genet 47:547–558
Wang YC, Ma H, Liu GF, Zhang DW, Ban QY, Zhang GD, Xu CX, Yang CP (2008) Generation and analysis of expressed sequence tags from a NaHCO3-treated Limonium bicolor cDNA library. Plant Physiol Biochem 46:977–986
Xiao YH, Li XB, Yang XY, Luo M, Hou L, Guo SH, Luo XY, Pei Y (2007) Cloning and characterization of a balsam pear class I chitinase gene (mcchit1) and its ectopic expression enhances fungal resistance in transgenic plants. Biosci Biotechnol Biochem 71:1211–1219
Ye XY, Ng TB (2005) A chitinase with antifungal activity from the mung bean. Protein Expr Purif 40:230–236
Acknowledgments
This work has been supported by Heilongjiang Province International Scientific and Technological Cooperation Project (WB07N02) and Natural Science Foundation of Heilongjiang Province (QC07C56).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Liu, Z.H., Yang, C.P., Qi, X.T. et al. Cloning, Heterologous Expression, and Functional Characterization of a Chitinase Gene, Lbchi32, from Limonium bicolor . Biochem Genet 48, 669–679 (2010). https://doi.org/10.1007/s10528-010-9348-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10528-010-9348-x