
Abstract
A gene encoding a novel chitinase chi58 was cloned from the fungus Chaetomium cupreum by using inverse PCR. The DNA sequence of chi58 contains a 1,602 bp open reading frame and two introns that are 52 and 201 bp in length. Regarding our in silico analysis, chi58 is a modular enzyme composed of a family-18 catalytic domain, which is responsible for chitinase activity, and a chitin-binding domain containing several cysteines. Apparently, the function of these domains is to anchor the enzyme tightly onto the large insoluble polymeric substrate. Chi58 has a pI of 4.47 and a deduced molecular mass of 58 kDa. The optimal pH and temperature conditions were determined to be 5.8 and 45°C, respectively, when colloidal chitin was used as the substrate. SDS-PAGE and zymogram analyses indicated the presence of a single active chitinase. Cells with pPIC9K-chi58 produced an extracellular chitinase that had an activity of 39 U/ml protein. Metal ions such as Ba2+, Mg2+, K+, Cu2+, Fe3+, Zn2+, and Co2+ also influenced the activity of the recombinant enzyme.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Chaetomium cupreum is a widely distributed soil fungus that antagonizes numerous fungal phytopathogens (Yang 2003). The antagonism of C. cupreum is usually correlated with antifungal activities, including the secretion of cell-wall-degrading enzymes such as chitinases (EC 3.2.1.14) (Inglis and Kawchuk 2002). Chitinases catalyze hydrolytic cleavage of the β-1,4-glycoside bonds present in N-acetyl-glucosamine biopolymers, mainly chitin (Cohen-Kupiec and Chet 1998). Thus, chitinases that hydrolyze chitin-containing fungal cell walls are thought to play a major role in the defense response against phytopathogens. Microbial chitinases are attractive as candidates for the control of phytopathogenic fungi and insect pests (Roco and Prez 2001). Chitin-hydrolyzing enzymes are classified into three categories [endochitinases, exochitinases (EC 3.2.1.14), and N-acetylglucosaminases] based on the manner in which they cleave the chitin chains. Endochitinases randomly cleave the β-1,4-glycosidic bonds of chitin, whereas exochitinases cleave the chain from the nonreducing end to form diacetyl-chitobiose (GlcNAc2). N-acetylglucosaminases hydrolyze GlcNAc2 to GlcNAc or produce GlcNAc from the nonreducing end of N-acetyl-chitooligosaccharides (Tanaka et al. 2001). The chitinases that have been sequenced so far are classified into two different families based on the amino acid sequence similarity of their catalytic domains. These are families 18 and 19 in the family classification system of glycoside hydrolases (Henrissat 1991). Family 18 contains chitinases from bacteria, fungi, viruses, and animals together with some plant chitinases. On the other hand, family 19 contains only plant chitinases and Streptomyces griseus chitinase (Ohno et al. 1996).
To date, various chitinases have been isolated from biocontrol fungi such as Trichoderma atroviride (Kubicek et al. 2001), Trichoderma harzianum (Shakeri and Foster 2007), and Chaetomium globosum (Liu et al. 2008). No chitinase gene has been cloned from C. cupreum, and very little is known regarding high-level expression and characterization of chitinase genes from Chaetomium spp. The chitin-binding domain (ChBD) is considered to play an important role in the biocontrol activity of this strain against phytopathogens (Kitamura and Kamei 2003). Limón et al. (2004) have confirmed the importance of endochitinases in the antagonistic activity of T. harzianum strains and demonstrated the effectiveness of adding a ChBD to increase the hydrolytic activity toward insoluble substrates such as chitin-rich fungal cell walls.
The methylotrophic yeast Pichia pastoris has been widely and successfully used for high-level expression of heterologous proteins from various sources such as bacteria, fungi, plants, viruses, invertebrates, vertebrates, and humans. Use of a P. pastoris expression system with an α-factor signal sequence of Saccharomyces cerevisiae, a c-myc epitope, and 6 His codons added at the 3′-end of the target genes led to an increase in the production of recombinant proteins to more than 30% of the total protein content (Labarre et al. 2007). In this study, we describe the cloning and expression of a C. cupreum chitinase gene in P. pastoris. The gene was expressed using the pPIC9K vector containing the AOX1-methanol inducible promoter. Functional expression in P. pastoris will facilitate large-scale production of this enzyme, which would allow its use in biocontrol and industrial applications and permit structural and functional studies.
Materials and Methods
Fungal Strains, Media, and Culture Conditions
The C. cupreum strain was kindly provided by King Mongkut’s Institute of Technology, Ladkrabang, Thailand. Chi58 was cloned in Escherichia coli JM109 cells. Cells carrying the recombinant plasmids were grown in Luria-Bertani (LB) medium containing 100 μg/ml ampicillin. The P. pastoris strain (Invitrogen, Carlsbad, CA) GS115 (Mut+/His+) was used as the host for expressing C. cupreum chi58. Chitin, carboxymethyl chitosan, and glycol chitosan were purchased from Sigma Chemical Co. (St. Louis, MO).
Genomic DNA Isolation
A piece of C. cupreum mycelium from an agar slant was inoculated onto potato dextrose medium and incubated at 28°C for 48 h with continuous shaking at 180 rpm. The mycelia were harvested and stored at −70°C for DNA extraction. DNA was extracted using the CTAB method described previously (Zhang et al. 1996).
Inverse PCR (IPCR) Amplification of chi58
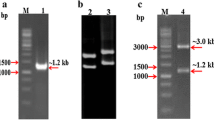
We cloned the chi58 gene from C. cupreum using an IPCR method that employed nested primers directed against a chitinase-encoding EST (GenBank accession no. DV546055) extracted from the C. cupreum mycelium cDNA library. The IPCR reaction was subjected to two-step PCR (nested PCR). The reaction mixture for the first PCR (50 μl) contained 5 μl 10× buffer, 2 μl 10 mmol/l dNTP, 2.5 μl primers (0.2 mmol/l each primer), 1 μl 5 U/μl Ex Taq, 5 μl (10 ng/μl) genomic DNA, and 32 μl ddH2O. The sense primer p1 was 5′-GTGTCTTTGTCTCTGTCGTGGC-3′, and the antisense primer p2 was 5′-ACGTCTCGGCTATCAAGAAC-3′. The reaction mixtures were incubated at 94°C for 5 min, followed by 35 cycles of denaturation (94°C for 30 s), annealing (55°C for 30 s), and extension (72°C for 3 min). A final extension was carried out at 72°C for 10 min. Using the reaction products (2 μl) of the first PCR as the template DNA, the second PCR was performed with the p3 and p4 primer pair (5′-GACGAGGAAACTAGACTCATAG-3′ and 5′-CTGGTCACTTTCATGATGG-3′). Amplification was carried out under the following conditions: 35 cycles of denaturation at 94°C for 30 s, annealing at 60°C for 30 s, and extension at 72°C for 3 min.
Total RNA Extraction and RT-PCR
For chitinase expression studies, C. cupreum was first precultured for 48 h on a potato dextrose medium. The mycelia were collected by filtration, washed three times with sterilized water, and transferred to a liquid synthetic medium, as described previously (Robertus and Monzingo 1999). The carbon sources in the synthetic medium were glucose and colloidal chitin. After 48 h of incubation, the mycelia were harvested for RNA extraction.
Total RNA was isolated using the Plant RNA extractive kit (Invitrogen Biotechnology, Shanghai). RNA integrity was confirmed by running samples on a 0.8% agarose gel. RT-PCR amplification was performed with the RNA PCR Kit (AMV) (TaKaRa, Japan), according to the manufacturer’s protocol.
Construction of Recombinant pPIC9K-chi58
cDNA fragments encoding chi58 were cloned into the P. pastoris expression vector pPIC9K in-frame to the pPIC9K N-terminal secretory signal peptide in order to produce recombinant fusion proteins that would be secreted into the growth medium. The following primers were used to amplify the chi58 gene ORF: chiL 5′-GACCGGAATTCAGCACGAGGCAAAAGCTC-3′ (the EcoRI site is italicized) and chiR 5′-CCAGCGGCCGCTTAACTGCTTCCTGAATCGATGT-3′ (the NotI site is italicized). The ORF of the chi58 gene was amplified with the chiL and chiR primers, digested with EcoRI and NotI, ligated into the expression vector of pPIC9K (Invitrogen), and transferred in E. coli JM109 cells for amplification.
Transformation and Expression in P. pastoris
Following linearization with SalI, each vector was used to transform P. pastoris strain GS115 by means of electroporation. This was carried out in a gene pulser apparatus (Bio-Rad) using high voltage pulses of 1100 V/0.6 mA in 0.1 cm electrode gap cuvettes. P. pastoris GS115 competent cells (80 μl volume) were transformed with 5 μg linearized DNA, and the transformed cells were rapidly diluted with 1 ml ice-cold 1 mol/l sorbitol, followed by plating on MD plates. The plates were incubated at 30°C for 3–4 days.
A single colony of the recombinant yeast strain (His+/Mut+) was inoculated in 100 ml of BMGY medium and grown at 28–30°C in a shaking incubator (250–300 rpm) until the culture reached OD600 = 2–6 (approximately 16–18 h). The cells were harvested by centrifugation at 1,500–3,000g for 5 min at room temperature. To induce expression, the cell pellets were resuspended in BMMY medium after 24 h until the broth reached OD600 = 5–10. Methanol (100%) was added to a final concentration of 0.5% every 24 h to maintain induction, and the cells were finally harvested.
Measurement of Enzyme Activity
Chitinase activity was measured according to the Schales procedure (Imoto and Yagishita 1971) but with some modifications. The reaction mixture, consisting of 0.5 ml colloidal chitin (1%, w/v) as the substrate and 0.5 ml enzyme solution (in 0.02 mol/l HAc–NaAc buffer, pH 4.5), was incubated at 37°C for 20 min, boiled for 5 min, supplemented with 1 ml 0.05% (w/v) KFe(CN), and finally boiled again for 10 min. After cooling, the reducing sugars that were released as a result of chitinase activity were measured at 420 nm. One unit of chitinase activity was defined as the amount of enzyme that produced 1 μg of reducing N-acetyl-d-glucosamine per hour. Colloidal chitin was obtained by the method of Roberts and Selitrennikoff (1988). Glycol chitin was obtained by acetylation of glycol chitosan (Trudel and Asselin 1989).
SDS-PAGE and Zymogram Analyses of Chitinase
Sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) was carried out by the method of Laemmli (1970). After SDS-PAGE, the gel was stained with 0.05% Coomassie brilliant blue R-250. Zymogram analysis was performed as follows (Trudel and Asselin 1989). Protein samples were added to the SDS-PAGE sample buffer and heated at 100°C for 5 min. The proteins were separated on a 12% polyacrylamide gel containing 0.1% glycol chitin, and the gel was immersed in refolding buffer (50 mmol/l Tris–HCl, pH 7.5; 1% Triton X-100) at 37°C overnight. The gel was washed with distilled water and then stained with 0.01% (w/v) Calcofluor white M2R in 10 mmol/l Tris–HCl (pH 7.5). After 5 min, the brightener solution was removed, and the gel was washed with distilled water. Lytic zones were visualized by placing the gels on a UV transilluminator.
Results
Cloning and Sequence Analysis of the Chitinase Gene
By screening 3,066 clones from a C. cupreum cDNA library, a chitinase-encoding EST (DV546055) was identified by BlastX analysis. Based on the cDNA sequence, a pair of nested primers was designed to perform IPCR on genomic DNA in order to amplify the full-length DNA sequence. The DNA sequence of chi58 is 2,065 bp in length and contains a 1,602 bp open reading frame and two introns that are 52 bp and 201 bp in length. Chi58 encodes a protein of 533 amino acids with a calculated molecular weight of 58 kDa and a pI of 4.47. The nucleotide sequence was registered in the GenBank nucleotide sequence database with accession no. DQ886936. In silico analysis showed that the sequence has high homology with the chitinases encoded by C. globosum (74%), Aspergillus clavatus (54%), Aspergillus nidulans (53%), and Neosartorya fischeri (49%). The encoded protein contains a putative signal peptide with a predicted cleavage site located between Ala17 and Ala18. The Ala-X-Ala sequence is frequently observed at the signal peptide processing sites of secretory proteins from bacteria and eukaryotes (von Heijne 1986). The location of four potential N-glycosylation sites (NXS/T) at Asn101, Asn139, Asn357, and Asn524 was also determined using the NetNGlyc version 1.0 program. In addition, 17 N-myristoylation sites, 14 casein kinase II phosphorylation sites, 6 protein kinase C phosphorylation sites, and 2 tyrosine sulfation sites were predicted by Prosite (http://www.expasy.org/cgi-bin/prosite).
Functional Domains of Chitinase
The CHI58 protein predicted by Smart (http://smart.embl-heidelberg.de) had a 9 amino acid chitinase family 18 active site (DXXDXDXE) (residues 259–267), which led to categorization of this protein as a member of glycosyl hydrolase family 18. A cysteine-rich linker sequence (residues 47–122) connected the catalytic domain to a 49 amino acid chitin-binding type-1 domain (residues 88–136). The chitin recognition or binding domain consisted of 20 amino acids (residues 105–124) (Fig. 1).

The structure of chi58 ORF from Chaetomium cupreum
CHI58 Activity and Substrate Specificity
Previous studies showed that chitinase production was low in S. cerevisiae (data not shown). Therefore, chi58 was cloned into the plasmid pPIC9K and subsequently transformed into P. pastoris strain GS115. The transformed GS115 clones secreted active chitinase enzyme into the liquid culture medium. When the transformant was cultured at 30°C for 120 h, maximum chitinase activity (39 U/ml) was obtained in the culture supernatant (Fig. 2).

Enzyme assays of pPIC9K-chi58. a Chitinase activity of recombinant pPIC9K-chi58 to induce expression for 6 days. b Effect of pH on the chitinase activity of recombinant pPIC9K-chi58. c Effect of temperature on the chitinase activity of recombinant pPIC9K-chi58. d Temperature stability of the recombinant pPIC9K-chi58. Maximum activity is set to 100%. The experiment was performed in triplicate
Chitinase exhibited maximum reactivity at pH 5.8 when colloidal chitin was used as the substrate (Fig. 2). When chitinase was assayed at different temperatures (35, 40, 45, 50, 55, 60, 70, and 80°C), maximum activity was observed at 45°C (Fig. 2). The thermal stability of chitinase was measured by incubating an aliquot of the enzyme at different temperatures for 10, 20, 30, 40, 50, and 60 min, and assaying the residual activity under optimal pH and temperature conditions (Fig. 2). Chitinase was stable at 40°C for 60 min and retained more than 90% of its initial activity. Incubation at 50°C for 60 min resulted in 65% residual activity. More than 50% of the maximal activity was retained when the enzyme was incubated for at least 50 min at 60°C; however, the residual activity of the enzyme was completely lost on incubation at 70°C. The enzyme could not hydrolyze glycol chitosan but could hydrolyze glycol chitin, colloidal chitin, and soluble chitosan (Table 1).
Effects of Metal Ions and Chemicals on Enzyme Activity
Table 2 shows the effects of metal ions and chemicals on chitinase activity. Among the various metal ions assayed, Ba2+, Mg2+, K+, and NH4 + ions (each at 5 mmol/l) enhanced the enzyme activity by 171.2%, 21.2%, 24.4%, and 80%, respectively. In contrast, Fe3+, Cu2+, Zn2+, Co2+ (each at 5 mmol/l), and SDS (0.5%) reduced the enzyme activity by 51.1%, 52.2%, 52.8%, 43.1%, and 60.6%, respectively. The degree of activation and inhibition of enzyme activity is expressed as a percentage of the enzyme activity in the control sample (no additional metal ion or chemical agent present).
Expression and Characterization of CHI58 in P. pastoris
SDS-PAGE showed an apparently new band at 58 kDa (Fig. 3), consistent with the theoretically predicted molecular weight. The maximum expression yield was observed at 120 h after induction with methanol. The presence of only one band on the zymogram of chitinase on a native gel indicated the presence of a single active chitinase (Fig. 3).

SDS-PAGE analysis of samples of expression protein (a). Samples were resolved on 12% polyacrylamide gel and then stained with Coomassie Blue R-250. Lane 1, protein marker (116, 66, 45, 31, 20.1, 14.4 kDa). Lanes 2–4, expression of pPIC9K induced with 0.5% (v/v) methanol at 24, 72, and 120 h. Lanes 5–9, expression of pPIC9K-chi58 induced with 0.5% (v/v) methanol at 24, 48, 72, 96, and 120 h. Arrows show induced chitinase, molecular weight of 58 kDa. Zymogram of pPIC9K-chi58 (b). Sample induced at 120 h was used for zymogram analysis
Discussion
In this study, we cloned the C. cupreum chitinase gene chi58. After reviewing the existing literature, we believe that this is the first report on the cloning of the chi58 gene from the biocontrol fungus C. cupreum. Sequence analysis showed that this gene is a typical member of glycosyl hydrolase family 18. The CHI58 polypeptide includes a signal sequence at the N-terminal end, suggesting that it is a secreted enzyme. Multiple sequence alignments indicated that CHI58 contains a ChBD “CPLXVCCSXXGFCGTXXXFC” (Fig. 4), which is unusual for fungal enzymes. Amino acid alignment revealed that this chitinase has a cysteine-rich region. Recently, the ChBD has been regarded as distinct from the cellulose binding domain, and the former has been classified into types 1, 2, and 3 (http://www.ncbi.nlm.nih.gov/Structure/cdd/cdd.shtml), found in ChBD from fungi and plants, viruses and insects, and bacteria, respectively. The ChBD of chitinases is similar to the domain found in plant chitinases, which contains conserved cysteine residues. In contrast, several aromatic amino acids, principally tryptophan, are prominent among the residues conserved in the bacterial ChBD. Some chitinases from plants and other proteins with no chitinase activity (lectins from plants and peritrophins from insects) also have ChBDs containing several cysteines, but these domains are generally located near the N- or C-terminal ends of the proteins (Blaxter 1996). Apparently, the function of these domains is to anchor the enzyme tightly onto the large insoluble polymeric substrate.

Comparison of predicted amino acid sequence for chitinase identified from Chaetomium cupreum (Cc) with chitinase from C. globosum (Cg; GenBank acc. no. XM001230073), Aspergillus niger (An; XM001396587), Magnaporthe grisea (Mg; XM362089), A. terreus (At; XM001209977), and A. clavatus (Ac; XM001269011). The chitin recognition or binding domain is marked by a black box. Asterisks (*) below the amino acid sequences indicate identical, highly conserved. Alignments were performed by the ClustalX method using the DNAman program
The results showed that CHI58 is active over a broad pH range (5.0–8.0), with the optimum being at 5.8. Similar pH optima have been reported for other chitinases. For example, pH 5.4 was reported as the optimum value for CHIT60 from Serratia plymuthica HRO-C48 (Jens et al. 2001); pH 5.5, Bacillus sp. WY22 (Woo and Park 2003); pH 5.5, Enterobacter sp. NRG4 (Lee et al. 2007); and pH 6.0, Enterobacter sp. G-1 (Park et al. 1997). Chitinases from different sources can use a variety of substrates. In this study, CHI58 could hydrolyze colloidal chitin, powdered chitin, carboxymethyl chitosan, and glycol chitin but failed to degrade glycol chitosan. These results are similar to those reported earlier (Lee et al. 2007).
Previous publications reported that EDTA was an inhibitor of chitinase, i.e., it inhibited the chitinase from Enterobacter sp. NRG4 (Neetu et al. 2005) and an exochitinase from Bacillus thuringiensis subsp. (de la Vega et al. 2006). In this study, addition of EDTA at a similar concentration (5 mmol/l) had little inhibitory activity, suggesting that these chitinases may have different catalytic mechanisms. CHI58 is a metalloenzyme, and chitinases from different fungi exhibit different responses to various metal ions. For example, the activity of the chitinase from Alcaligenes xylosoxydans was inhibited by 25% when 5 mmol/l Cu2+ was present but was unaffected by the same concentration of Ca2+, Ba2+, or Mg2+ (Vaidya et al. 2003). Enterobacter sp. G-1 chitinase activity was unaffected by Ca2+ addition to the enzyme solution (Park et al. 1997). The chitinase from Enterobacter aerogenes was stimulated by Zn2+, Ba2+, Ca2+, and Mn2+ and strongly inhibited by Co2+ and Mg2+ (Tang et al. 2001). The chitinase from Bacillus sp. DAU101 was strongly inhibited by Zn2+, Cu2+, and EDTA. However, the enzyme activity increased when Co2+, Mg2+, Ca2+, or Ba2+ was added (Lee et al. 2007). In this study, enhancement of chitinase activity by Ba2+, Mg2+, K+, and NH4 + ions (each at 5 mmol/l) was 171.2%, 21.2%, 24.4%, and 80%, respectively. These cations probably protect the enzyme against thermal denaturation and therefore maintain the active conformation of the enzyme at high temperatures (Donaghy and Mckay 1993).
The methylotrophic yeast P. pastoris is a useful host for the production of a variety of prokaryotic and eukaryotic proteins (Cereghino and Cregg 2000). For example, the yield of chitinase Bbchit1 from Beauveria bassiana produced by P. pastoris GS115 was 153 mg/l, which was significantly higher than that obtained by refolding this protein from E. coli inclusion bodies (50 mg/l). Additionally, the specific activity of Bbchit1 from P. pastoris was also higher than that from E. coli (Fitches et al. 2004; Fan et al. 2007). CHI58 expressed in P. pastoris was largely released into the extracellular medium, and zymogram analysis indicated the presence of a single active chitinase. Our results and those of Fan et al. (2007) demonstrate that P. pastoris GS115 is a suitable strain for the expression of foreign chitinase genes.
Finally, high-level CHI58 expression may be possible by codon optimization. Detailed structural and functional analyses of the chitinase, especially its interaction with pathogenic fungi, can be carried out by means of site-directed mutagenesis or other genetic techniques. Protein engineering in such systems would allow the rational design of mutants with improved properties. Chitinase production in P. pastoris will facilitate large-scale protein production and allow structural and functional studies on chitinase. Moreover, the methods described here will be helpful in the detection, identification, and cloning of chitinases from uncultured/unknown strains as well as in the characterization of other genes.
References
Blaxter M (1996) Protein motifs in filarial chitinases. Parasitol Today 12:42
Cereghino JL, Cregg JM (2000) Heterologous protein expression in the methylotrophic yeast Pichia pastoris. FEMS Microbiol Rev 24:45–66
Cohen-Kupiec R, Chet I (1998) The molecular biology of chitin digestion. Curr Opin Biotechnol 9:270–277
de la Vega LM, Barboza-Corona JE, Aguilar-Uscanga MG, Ramírez-Lepe M (2006) Purification and characterization of an exochitinase from Bacillus thuringiensis subsp. aizawai and its action against phytopathogenic fungi. Can J Microbiol 52:651–657
Donaghy JA, Mckay AM (1993) Production and properties of an alkaline protease by Aureobasidium pullulans. J Appl Bacteriol 74:662–666
Fan YH, Zhang YJ, Yang XY, Pei XQ, Guo SJ, Pei Y (2007) Expression of a Beauveria bassiana chitinase (Bbchit1) in Escherichia coli and Pichia pastoris. Protein Expr Purif 56:93–99
Fitches E, Wilkinson H, Bell H, Bown DP, Gatehouse JA, Edwards JP (2004) Cloning, expression and functional characterization of chitinase from larvae of tomato moth (Lacanobia oleracea): a demonstration of the insecticidal activity of insect chitinase. Insect Biochem Mol Biol 34:1037–1050
Henrissat B (1991) A classification of glycosyl hydrolases based on amino acid sequence similarities. Biochem J 280:309–316
Imoto T, Yagishita K (1971) A simple activity measurement of lysozyme. Agric Biol Chem 35:1154–1156
Inglis GD, Kawchuk LM (2002) Comparative degradation of oomycete, ascomycete, and basidiomycete cell walls by mycoparasitic and biocontrol fungi. Can J Microbiol 48:60–70
Jens F, Matteo L, Felice S, Roland S, Gabriele B, Hubert B (2001) Purification and properties of two chitinolytic enzymes of Serratia plymuthica HRO-C48. Arch Microbiol 176:421–426
Kitamura E, Kamei Y (2003) Molecular cloning, sequencing and expression of the gene encoding a novel chitinase A from a marine bacterium, Pseudomonas sp. PE2, and its domain structure. Appl Microbiol Biotechnol 61:140–149
Kubicek CP, Mach RL, Peterbauer CK, Lorito M (2001) Trichoderma: from genes to biocontrol. J Plant Pathol 83:11–23
Labarre C, van Tilbeurgh H, Blondeau K (2007) Pichia pastoris is a valuable host for the expression of genes encoding membrane proteins from the hyperthermophilic Archeon Pyrococcus abyssi. Extremophiles 11:403–413
Laemmli UK (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685
Lee YS, Park IH, Yoo JS, Chung SY, Lee YC, Cho YS, Ahn SC, Kim CM, Choi YL (2007) Cloning, purification, and characterization of chitinase from Bacillus sp. DAU101. Bioresour Technol 98:2734–2741
Limón MC, Chacón MR, Mejías R, Delgado-Jarana J, Rincón AM, Codón AC, Benítez T (2004) Increased antifungal and chitinase specific activities of Trichoderma harzianum CECT 2413 by addition of a cellulose binding domain. Appl Microbiol Biotechnol 64:675–685
Liu ZH, Yang Q, Hu S, Zhang JD, Ma J (2008) Cloning and characterization of a novel chitinase gene (chi46) from Chaetomium globosum and identification of its biological activity. Appl Microbiol Biotechnol 80:241–252
Neetu D, Rupinder T, Ram PT, Gurinder SH (2005) Chitinase from Enterobacter sp. NRG4: its purification, characterization and reaction pattern [Online]. Electron J Biotechnol 8(2). Available from http://www.ejbiotechnology.info/content/vol8/issue2/full/6/index.html. ISSN: 0717–3458
Ohno T, Armand S, Hata T, Mikaidou N, Henrissat B, Mitsutomi M, Watanabe T (1996) A modular family 19 chitinase found in the prokaryotic organism Streptomyces griseus HUT 6037. J Bacteriol 178:5065–5070
Park JK, Kenji M, Ikuo F, Yukikazu Y, Tsuyoshi N, Makoto K, Hideyuki M (1997) Purification and characterization of the chitinase (ChiA) from Enterobacter sp. G-1. Biosci Biotechnol Biochem 61:684–689
Roberts WK, Selitrennikoff CP (1988) Plant and bacterial chitinases differ in antifungal activity. J Gen Microbiol 134:169–176
Robertus JD, Monzingo AF (1999) The structure and action of chitinases. EXS 87:125–135
Roco A, Prez LM (2001) In vitro biocontrol activity of Trichoderma harzianum on Alternaria alternata in the presence of growth regulators [Online]. Electron J Biotechnol 4(2). Available from http://www.ejbiotechnology.info/content/vol2/issue3/full/3/index.html. ISSN: 0717–3458
Shakeri J, Foster HA (2007) Proteolytic activity and antibiotic production by Trichoderma harzianum in relation to pathogenicity to insects. Enzyme Microb Technol 40:961–966
Tanaka T, Fukui T, Imanaka T (2001) Different cleavage specificities of the dual catalytic domains in chitinase from the hyperthermophilic archaeon Thermococcus kodakaraensis KOD1. J Biol Chem 276:35629–35635
Tang Y, Zhao J, Ding S, Liu S, Yang Z (2001) Purification and properties of chitinase from Enterobacter aerogenes. Wei Sheng Wu Xue Bao (Acta Microbiol Sin) 41:82
Trudel J, Asselin A (1989) Detection of chitinase activity after polyacrylamide gel electrophoresis. Anal Biochem 178:362–366
Vaidya R, Roy S, Macmil S, Gandhi S, Vyas P, Chhatpar HS (2003) Purification and characterization of chitinase from Alcaligenes xylosoxydans. Biotechnol Lett 25:715–717
von Heijne G (1986) A new method for predicting signal sequence cleavage sites. Nucleic Acids Res 14:4683–4690
Woo CJ, Park HD (2003) An extracellular Bacillus sp. chitinase for the production of chitotriose as a major chitinolytic product. Biotechnol Lett 25:409–412
Yang Q (2003) Evaluation of Chaetomium for biological control of Fusarium wilt of tomato in P. R. China. In: Yang Q (ed) Biological control and biotechnology. Heilongjiang Science and Technology Press, Harbin, pp 25–36
Zhang D, Yang Y, Castlebury LA, Cerniglia CE (1996) A method for the large scale isolation of high transformation efficiency fungal genomic DNA. FEMS Microbiol Lett 145:216–265
Acknowledgments
This study was supported by the Chinese National Programs for High Technology Research and Development (2003AA241140).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Wang, YJ., Yang, Q. Cloning and Expression of a Novel Chitinase chi58 from Chaetomium cupreum in Pichia pastoris . Biochem Genet 47, 547–558 (2009). https://doi.org/10.1007/s10528-009-9251-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10528-009-9251-5