
Abstract
Bitter taste reception is expected to be associated with dietary selection and to prevent animals from ingesting potentially harmful compounds. To investigate the genetic basis of bitter taste reception, we reconfirmed the bitter taste receptor (T2R) genes from cow (herbivore) and dog (carnivore) genome sequences and identified the T2R repertoire from the draft genome of the bat (insectivore) for the first time using an automatic data-mining method. We detected 28 bitter receptor genes from the bat genome, including 9 intact genes, 8 partial but putative functional genes, and 9 pseudogenes. In the phylogenetic analysis, most of the T2R genes from the three species intermingle across the tree, suggesting that some are conserved among mammals with different dietary preferences. Furthermore, one clade of bat-specific genes was detected, possibly implying that the insectivorous mammal could recognize some species-specific bitter tastants. Evolutionary analysis shows strong positive selection was imposed on this bat-specific cluster, indicating that positive selection drives the functional divergence and specialization of the bat bitter taste receptors to adapt diets to the external environment.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Bitter taste receptor (T2R) genes have been considered as bitter taste receptors in mammals, belonging to the G protein-coupled receptors characterized by seven transmembrane regions and highly conserved amino acid residues (Chandrashekar et al. 2000; Glendinning 1994; Glendinning et al. 1999; Wong et al. 1996). About 900 base pairs (bp) long, the T2R genes are intronless, showing a very simple gene structure (Adler et al. 2000). For this reason, T2R repertoires have been examined from an array of mammals, including humans (Fischer et al. 2005; Shi et al. 2003), mice (Shi et al. 2003), rats (Shi and Zhang 2006), dogs, cattle, and opossums (Go 2006; Shi and Zhang 2006), using data-mining approaches against genome sequences. It is well known that natural poisonous substances generally taste bitter (Chandrashekar et al. 2000; Glendinning and Smith 1994; Glendinning et al. 1999); thus, the reception of bitter tastes may play a role in the survival of mammals. Actually, animals may encounter different bitter compounds depending on dietary ingredients and ecological niches. To date, the bitter taste receptor genes have been investigated from omnivora (human, mouse, rat, and opossum), herbivore (cow), and carnivore (dog), but nothing is known about T2R genes in insectivorous mammals.
Bats (Chiroptera) are distributed widely across the globe. As the only mammals with true flight, they occupy a nocturnal sky (Simmons 2005). Traditionally bats have been divided into Megachiroptera (megabats) and Microchiroptera (microbats). Megabats feed on fruit, pollen, and nectar without echolocation (the only exception being the genus Rousettus), whereas bats of the Microchiroptera, such as the little brown bat (Myotis lucifugus), are sophisticated echolocators preying mostly on insects (Patterson et al. 2003). The little brown bat, distributed in America, Canada, and Mexico, consumes mainly aquatic insects using frequency modulated calls (Fenton and Barclay 1980; Fenton and Bell 1979). The availability of the draft genome sequence of Myotis lucifugus provided an opportunity to identify the bat T2R repertoire, which is expected to shed light on the evolutionary mechanism of bitter tastes and the adaptation for bat survival. To address this issue, we first report the identification of T2R genes from bats and then conduct an evolutionary analysis of these genes.
Materials and Methods
Data
The draft genome sequence of the little brown bat (Myotis lucifugus; released in May 2006; 1.7× coverage) was downloaded from NCBI (http://www.ncbi.nlm.nih.gov/), with GenBank accession number AAPE00000000. Sequences from the cow and dog were retrieved from the Ensembl database: cow (7.1× coverage), dog (7.6× coverage).
Detection of T2R Genes
First, we collected representative T2R genes identified from humans, mice, rats, chickens, and frogs as query T2R sequences. Then, we performed TBlastN (Altschul et al. 1997) searches against the little brown bat draft genome sequence with the E-value 1e−10. We used the nonoverlapping Blast hits to identify functional T2R genes. All Blast hit sequences were extended to both 3′ and 5′ directions along the genome scaffold sequences. The coding sequences containing the ATG and the stop codon were obtained and compared with the NCBI database using BlastP. If the best Blast hits of specific genes were not T2R genes, they were discarded. Some sequences missed the C-terminal or N-terminal portion and contained an initiation codon or stop codon. These genes were identified as partial T2R genes. The remaining sequences were used to estimate the nonsense or frame shift mutations using the profile HMM-based program Genewise2 (Birney et al. 2004).
Phylogenetic Tree and Evolutionary Analysis
The translated amino acid sequences of T2R genes in cattle, dogs, and bats were aligned using ClustalW (Thompson et al. 1994) with default parameters and evaluated by T-coffee (Notredame et al. 2000). The phylogenetic tree was constructed by Mega4 (Tamura et al. 2007) using the neighbor-joining method (Saitou and Nei 1987) with protein Poisson correction distances (Nei and Kumar 2000). The bootstrap values of the tree were evaluated with 1,000 replications (Felsenstein 1985).
A likelihood method was used to examine nucleotide substitutions by selective pressure using the Codeml program in the PAML software package (Yang 2007; Yang et al. 2000). The site-specific models were used to test possible positive selection (Wong et al. 1996; Yang et al. 2000, 2005). The models fix the ω ratios among branches but allow those among sites to vary. Here we used three pairs of site models. M0 assumes one ω ratio for all sites with only one site class, and M3 uses an unconstrained discrete distribution to model ω ratios with three site classes. M1a assumes two classes of sites, one constrained with 0 < ω0 < 1 and the other fixed with ω1 = 1; M2a, an extension of M1a, adds another class of sites, in which ω2 may be greater than 1 based on the data. M7 uses a beta distribution of the ω ratio, but with a constrained ω ratio smaller than 1; M8 adds an additional class of sites to M7, allowing some sites with a ω ratio greater than 1. Likelihood ratio tests (LRTs) were conducted to test positive selection (Yang et al. 2000). Here, we have three LRTs: M0 versus M3, M1a versus M2a, and M7 versus M8. Twice the log likelihood difference (2Δℓ) can be compared with a chi-square distribution; the degree of freedom is the difference of the numbers of parameters in the two models. LRT suggests positive selection if there is a significant difference between the two models by chi-square test.
Results
T2R Gene Repertoires in Bats, Cows, and Dogs
We used an automatic data-mining method to detect T2R genes in the little brown bat draft genome sequence. We ultimately detected a partial repertoire of little brown bat T2R genes, consisting of nine intact genes (900–996 bp), eight partial genes (357–876 bp), and nine pseudogenes (Table 1). The fraction of pseudogenes in the little brown bat draft genome sequence is ~35%. Considering that the current genome sequences of cows and dogs have a higher coverage, we also conducted searches against cow and dog genomes, identifying 18 functional genes and 15 pseudogenes from the cow genome and 15 functional genes and 5 pseudogenes from the dog genome (Table 1). All the amino acid sequences of intact T2R genes of bats, cows, and dogs are available in Supplementary Data.
Phylogenetic Analysis
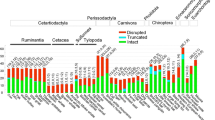
For the T2R genes of bats, some partial sequences are too short to make a good alignment. Therefore, we included only nine intact genes and four longer partial genes to make an alignment, together with all the functional genes of cows and dogs. Pseudogene sequences were removed from the phylogenetic analysis. The phylogenetic tree shows that T2R genes from the three mammals are mostly intermingled on the major clades (Fig. 1). This suggests that many gene duplication events occurred prior to the separation of cows, dogs, and bats; and these mammals may encounter some bitter compounds in common. Fortunately, there is a bat-specific cluster in the crown of this tree. The little brown bat is an insectivorous mammal, whereas cows are herbivorous and dogs are carnivorous. It is not surprising that this bat species would have some special bitter taste receptors that help it to avoid certain harmful substances in insects.

Neighbor-joining tree of 56 putatively functional T2R genes from the cow, dog, and bat. Numbers at interior nodes (≥50) are bootstrap values. Branch lengths are drawn to the scale (number of amino acid subsititutions per site). The dog V1R4 (GenBank accession no. NM_001013852) was used as outgroup but is not shown. Bat sequences are marked with a triangle; Bat_6T, Bat_8T, Bat_10T, and Bat_15T are partial sequences
Positive Selection of T2R Genes in a Bat-Specific Cluster
Our neighbor-joining tree of T2R genes contains a bat-specific cluster (Fig. 1). To explore the evolutionary forces on this cluster, we applied a maximum likelihood method. It is reported that positive selection may act on a few sites during protein evolution (Yang et al. 2000); therefore, the site-specific models available in the PAML package may have more power to detect positive selection. Here we conducted an analysis of models M0, M1a, M2a, M3, M7, and M8 using the Bat_8T, Bat_10T, Bat_T2R2, and Bat_T2R6 sequences as input data. After the removal of gaps, a total of 244 codons were used in the Codeml program. The average ω ratios (d N/d S), likelihood values, estimated parameters, and predicted positively selected sites are listed in Table 2. The models M3 and M0 can be compared using 4 degrees of freedom (M3 has 12 parameters, M0 assumes 8, 2Δℓ = 14.28). The chi-square test shows that the P value equals 0.006, suggesting that huge variation in selective pressure was present at amino acid sites. The same method is applied to the comparison of M1a versus M2a and M7 versus M8. The models M2a and M8 are significantly different from M1a and M7, indicating that the two models allowing positive selection fit our data significantly better than the null models. This is strong evidence of positive selection.
Adler et al. (2000) reported that T2Rs include three functional domains: transmembrane (TM), intracellular (IC), and extracellular (EC). The sites under selective pressure may be different in different domains. To examine the selective pressure on each functional domain, the inferred positively selected sites under model M3 were mapped (Fig. 2). Domains of TM1-6, IC1-3, and EC1-2, as well as the N-terminal, are shown, but EC3, TM7, and IC4 are missing in the low coverage of the bat genome. Although the data are incomplete, we still can get some information from this distribution. It is clear that there are more extracellular domains at possible positively selected amino acids than other regions (Fig. 2). The extracellular regions are the most divergent parts in the T2R genes, and possibly bind tastants (Adler et al. 2000; Gilbertson et al. 2000). This potential function may impose more selective pressure on ECs.

Posterior probabilities (>50%) for sites under positive selection. X-axis denotes position in the amino acid alignment. Y-axis denotes posterior probability of sites under positive selection. Boxes under the graph refer to T2R functional domains EC (extracellular), IC (intracellular), and TM (transmembrane) and to the N-terminal
Discussion
We have detected T2R genes from the draft genome sequence of the little brown bat. Because of the low coverage of the genome sequence, we identified only nine intact T2R genes and eight partial and putatively functional genes. We observed a bat-specific cluster in the phylogenetic tree reconstructed for cow, dog, and bat T2R genes. Our comparative evolutionary analysis of this clade and examination of the strong positive selection signatures suggest that adaptive diversification has occurred during the duplication events of the T2R genes.
Functional studies have shown that T2Rs respond to bitter tastants (Bufe et al. 2002; Chandrashekar et al. 2000), and bitter taste reception is expected to be associated with dietary selection. We chose three mammals with different dietary preferences to compare their T2R genes. Shi and Zhang (2006) did the analysis from cow and dog genome sequences, but we also updated these results in high-coverage genome sequences. Our results are almost identical to theirs, except that they identified an additional partial gene from cows and dogs. This slight difference could be attributed to different calculation methods. Because bitter compounds are considered more common in plant than in animal tissues, herbivores are expected to encounter more bitter substances than carnivores (Glendinning 1994). It is understandable that the number of functional T2R genes in cattle (18) is greater than in dogs (15), but the difference is not significant. Taking the highest proportion (45%) of pseudogenes into account, we speculate that cow bitter taste receptor genes might be relaxed from selective constraints due to the domestication event; humans can provide food to cows and protect them from poisonous compounds, so they reserve a lower number of functional T2R genes. Additionally, the rumination behavior of cows may reduce the risk from harmful foods. Considering dietary components and domestication, it is reasonable that the dog has a lower number of T2R functional genes than bats. Unfortunately, the bat genome sequence has very low coverage, and the number of putative T2R genes could be tentative, though we can gain some insight from the current incomplete data. It is interesting, however, that omnivorous mammals, such as humans, rats, mice, and opossums, have more functional T2R genes (Table 1). There is a good explanation: omnivora feed on both animal and plant tissues, thus they encounter more bitter tastants than do herbivora and carnivora (Shi and Zhang 2006).
The little brown bat has a specific cluster of T2R genes, suggesting that bats may have some unique bitter taste receptors. It is notable that bats occupy a nocturnal niche in the sky, an environment unique among mammals, which means that bats may encounter some specific bitter toxins. Thus, the specific gene cluster occurs during the early gene duplication events. Our maximum likelihood test suggests strong positive selection on the bat-specific cluster of T2R genes, and many sites with higher posterior probability are identified by site-specific models. Most sites affected by positive selection are distributed in the extracellular regions, which indicates that bitter tastants are selectively favored during bat evolution, as the extracellular domains are predictively involved in ligand binding (Adler et al. 2000; Gilbertson et al. 2000). Little brown bats are insectivorous, feeding on various species of insects, such as beetles, caddisflies, moths, mayflies, lacewings, and occasionally mosquitoes (Fenton and Barclay 1980; Fenton and Bell 1979). Many insects use chemical defenses for survival (Blum 1981; Eisner 1970; Weatherston and Percy 1970), which may increase the probability that insectivorous bats will encounter bitter substances. Positive selection may drive the functional divergence and specialization of bat bitter taste receptor genes and contribute to this bat’s ability to recognize a diversity of bitter tastants. This taste capability could protect these bats from harmful substances and thus be helpful to bat survival.
References
Adler E, Hoon MA, Mueller KL, Chandrashekar J, Ryba NJ, Zuker CS (2000) A novel family of mammalian taste receptors. Cell 100:693–702
Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25:3389–3402
Birney E, Clamp M, Durbin R (2004) GeneWise and Genomewise. Genome Res 14:988–995
Blum M (1981) Chemical defense of arthropods. Academic Press, New York
Bufe B, Hofmann T, Krautwurst D, Raguse JD, Meyerhof W (2002) The human TAS2R16 receptor mediates bitter taste in response to beta-glucopyranosides. Nat Genet 32:397–401
Chandrashekar J, Mueller KL, Hoon MA, Adler E, Feng L, Guo W, Zuker CS, Ryba NJ (2000) T2Rs function as bitter taste receptors. Cell 100:703–711
Eisner T (1970) Chemical defense against predators in arthropods. Academic Press, New York
Felsenstein J (1985) Confidence limits on phylogenies: an approach using the bootstrap. Evolution 39:783–791
Fenton M, Barclay R (1980) Myotis lucifugus. Mamm Species 142:1–8
Fenton M, Bell G (1979) Echolocation and feeding behaviour in four species of Myotis (Chiroptera). Can J Zool 57:1271–1277
Fischer A, Gilad Y, Man O, Paabo S (2005) Evolution of bitter taste receptors in humans and apes. Mol Biol Evol 22:432–436
Gilbertson TA, Damak S, Margolskee RF (2000) The molecular physiology of taste transduction. Curr Opin Neurobiol 10:519–527
Glendinning JI (1994) Is the bitter rejection response always adaptive? Physiol Behav 56:1217–1227
Glendinning JI, Smith JC (1994) Consistency of meal patterns in laboratory rats. Physiol Behav 56:7–16
Glendinning JI, Tarre M, Asaoka K (1999) Contribution of different bitter-sensitive taste cells to feeding inhibition in a caterpillar (Manduca sexta). Behav Neurosci 113:840–854
Go Y (2006) Proceedings of the SMBE Tri-National Young Investigators’ Workshop 2005. Lineage-specific expansions and contractions of the bitter taste receptor gene repertoire in vertebrates. Mol Biol Evol 23:964–972
Go Y, Satta Y, Takenaka O, Takahata N (2005) Lineage-specific loss of function of bitter taste receptor genes in humans and nonhuman primates. Genetics 170:313–326
Nei M, Kumar S (2000) Molecular evolution and phylogenetics. Oxford University Press, New York
Notredame C, Higgins DG, Heringa J (2000) T-Coffee: a novel method for fast and accurate multiple sequence alignment. J Mol Biol 302:205–217
Patterson B, Willig MR, Stevens RD (2003) Trophic strategies, niche partitioning, and patterns of ecological organization. University of Chicago press, Chicago
Saitou N, Nei M (1987) The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol 4:406–425
Shi P, Zhang J (2006) Contrasting modes of evolution between vertebrate sweet/umami receptor genes and bitter receptor genes. Mol Biol Evol 23:292–300
Shi P, Zhang J, Yang H, Zhang YP (2003) Adaptive diversification of bitter taste receptor genes in mammalian evolution. Mol Biol Evol 20:805–814
Simmons NB (2005) Mammal species of the world: a taxonomic and geographic reference. Johns Hopkins University Press, Baltimore, MD
Tamura K, Dudley J, Nei M, Kumar S (2007) MEGA4: molecular evolutionary genetics analysis (MEGA) software version 4.0. Mol Biol Evol 24:1596–1599
Thompson JD, Higgins DG, Gibson TJ (1994) Clustal W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 22:4673–4680
Weatherston J, Percy JE (1970) Arthropod defensive secretions. Academic Press, New York
Wong GT, Gannon KS, Margolskee RF (1996) Transduction of bitter and sweet taste by gustducin. Nature 381:796–800
Yang Z (2007) PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol 24:1586–1591
Yang Z, Nielsen R, Goldman N, Pedersen AM (2000) Codon-substitution models for heterogeneous selection pressure at amino acid sites. Genetics 155:431–449
Yang Z, Wong WS, Nielsen R (2005) Bayes empirical bayes inference of amino acid sites under positive selection. Mol Biol Evol 22:1107–1118
Acknowledgments
This work was supported by grants under the Key Construction Program of the National “985” Project and “211” Project.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Authors Yingying Zhou and Dong Dong contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Zhou, Y., Dong, D., Zhang, S. et al. Positive Selection Drives the Evolution of Bat Bitter Taste Receptor Genes. Biochem Genet 47, 207–215 (2009). https://doi.org/10.1007/s10528-008-9218-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10528-008-9218-y