
Abstract
Genetic risk for Late Onset Alzheimer Disease (AD) has been associated with lower cognition and smaller hippocampal volume in healthy young adults. However, whether these and other associations are present during childhood remains unclear. Using data from 5556 genomically-confirmed European ancestry youth who completed the baseline session of the ongoing the Adolescent Brain Cognitive DevelopmentSM Study (ABCD Study®), our phenome-wide association study estimating associations between four indices of genetic risk for late-onset AD (i.e., AD polygenic risk scores (PRS), APOE rs429358 genotype, AD PRS with the APOE region removed (ADPRS-APOE), and an interaction between ADPRS-APOE and APOE genotype) and 1687 psychosocial, behavioral, and neural phenotypes revealed no significant associations after correction for multiple testing (all ps > 0.0002; all pfdr > 0.07). These data suggest that AD genetic risk may not phenotypically manifest during middle-childhood or that effects are smaller than this sample is powered to detect.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Alzheimer Disease (AD) is a growing international public health problem. Alongside increases in global life expectancy (Wang et al. 2020), there have been increases in the AD and other dementia cases (117% increase from 1990 to 2016; (Nichols et al. 2019). This trend is expected to continue with projections of a 60% increase from 2019 to 2050 (from 57.4 to 152.8 million cases) due to three risk factors (i.e., high body-mass-index, fasting glucose, and smoking; GBD 2019 Dementia Forecasting Collaborators 2022). This anticipated increase in AD will generate further socioeconomic burden and negatively impact individuals, families, and health care (Grabher 2018; GBD 2019 Dementia Forecasting Collaborators 2022). As late-onset AD (LOAD) is largely heritable (58–79%) and characterized by an extensive polygenic architecture and a highly penetrant single common locus (i.e., APOE-ε4 Odds Ratio: 3.2; Sims et al. 2020; Mol et al. 2022), understanding the correlates of AD genetic risk across the lifespan may help identify and characterize early phenotypic signs, to ultimately improve our understanding of AD and limit its impact.
AD genetic risk is associated with variability in behavioral and neural phenotypes in healthy young adults
The typical late-life onset of (Late Onset) AD has led to efforts to identify precursors of the disorder that may enable early identification. Measures of cognition, brain structure, biomarkers (e.g., amyloid β), and genetic risk have been most frequently proposed and used to prognosticate later dementia and AD risk (Livingston et al. 2020). Mild cognitive impairment (MCI), which is characterized by memory complaints and/or impairment in the context of relatively preserved cognition, often precedes AD dementia (Albert et al. 2011). Similarly, hippocampal volume has been linked to reduced memory performance across ages, MCI, and progression to AD dementia (Filippini et al. 2009). Genetic risk (e.g., Apolipoprotein E genotype; polygenic risk scores) and measures of AD-related biomarkers (e.g. Amyloid β 42/40 ratio) have also been shown to predict the development of AD dementia (Bekris et al. 2010; Reitz et al. 2020; Bellenguez et al. 2022).
The investigation of AD genetic risk (i.e., APOE genotype, polygenic risk) among healthy individuals prior to the typical onset of AD dementia has revealed that variability in these cognitive and neural risk factors are observable in healthy young adults as early as in their thirties (Hendriks et al. 2021). For example, genetic risk for AD (i.e., APOE genotype, polygenic risk) has been associated with smaller hippocampal volume and lower cognition across many studies of young (mean age = 26.8), middle (ages 45–55), and older aged adults (mean age = 72) (Fleisher et al. 2005; O’Dwyer et al. 2012; Evans et al. 2020; Walhovd et al. 2020; Murray et al. 2021). These data suggest that subtle differences in cognition and brain structure are present even before onset of clinical impairment. What remains unclear is whether these differences emerge during childhood and if AD genetic risk is associated with factors beyond cognition and hippocampal volume including other behavioral, neural, experiential, and social factors (Dean et al. 2014; Korologou-Linden et al. 2022).
The current study
Here, we conducted a phenome-wide association study (PheWAS) of genetic risk for late-onset Alzheimer Disease among children of European ancestry who completed the baseline session of the Adolescent Brain and Cognitive DevelopmentSM (ABCD) Study. We hypothesized that genetic risk for AD (e.g., APOE rs429358 risk allele and AD polygenic risk) would be associated with smaller hippocampal volume and lower cognitive performance during middle childhood, and that novel associations with behavioral and brain phenotypes and experiential and social factors would be identified.
Methods
Participants
Data were drawn from data release 3.0 and 4.0 of the ongoing longitudinal Adolescent Brain and Cognitive DevelopmentSM (ABCD) Study (Volkow et al. 2018). The ABCD Study® is following 11,879 children (ages 8.9–11) recruited at baseline from 22 research sites across the United States to study the development of complex behavior and biology during middle childhood to late adolescence/young adulthood in the context of experience and genetic background. We drew data only from the baseline session. Participants of non-European genomic ancestry were excluded (see “Genetic Data” section below) from analyses due to the lack of a well powered ancestry-specific discovery GWAS of Alzheimer Disease in other ancestries, the relatively uninformative and low predictive utility of PRS when applied across ancestries (Martin et al. 2019), and evidence of divergence in genetic risk for AD across ancestries (Kunkle et al. 2021). After further excluding individuals with missing covariate data, our final analytic sample consisted of 5,556 children of genomically-confirmed European ancestry with baseline study data. Analytic Ns ranged from 120 to 5556 (mean N = 5012; median N = 5509) due to missing phenotypic data.
Phenotypes
All ABCD Study baseline behavioral, self-report, and neuroimaging phenotype data (data release 3.0 and 4.0) as well as genomic data (data release 3.0) were downloaded from the National Institute of Mental Health Data Archive (NDA; https://nda.nih.gov/). Phenotypes were reviewed for inclusion according to: (1) relevance (e.g., administrative items [e.g., measurement device], redundancy [e.g., excluding t-scored data and using raw data]; Supplement), and (2) missingness and frequency variability (i.e., continuous phenotypes were required to have ≥ 100 participants with non-missing values; categorical variables required ≥ 100 endorsements/category). When applicable (e.g., substance use questions that were not asked following a response that the child had not heard of the substance), missing data were recoded to 0. Otherwise, all missing values were coded as missing (e.g., distress related to the presence of psychotic-like experiences was coded as missing in participants who reported no psychotic-like experiences). All data were triple checked by multiple investigators for relevance, variability, and accurate recoding.
Data were separated into those corresponding to behavioral and psychosocial phenotypes (N = 1269; Supplementary Tables 1 and 3) and brain imaging phenotypes (N = 418; Supplementary Table 2) for analyses (see “Statistical Analyses”). Behavioral and psychosocial data were categorized into the following 8 domains: (1) cognition (N = 14), (2) screen time (N = 18), (3) demographics (N = 27), (4) substance (N = 48), (5) culture/environment (N = 113), (6) physical health (N = 170), (7) family mental health (N = 239), and (8) child mental health (N = 640) (Supplementary Table 3). Neuroimaging indices of brain structure and resting state functional connectivity (RSFC) included the following domains (processing details provided in the Supplement): (1) gray matter volumes (global N = 9; subcortical N = 35, cortical N = 68; Supplementary Tables 5–6), (2) cortical thickness (global N = 3; regional N = 68; Supplementary Table 5–6), (3) cortical surface area (global N = 3; regional N = 68; Supplementary Table 5–6), (4) DTI fractional anisotropy (global N = 1; N = 37; Supplementary Table 5–6), (5) DTI mean diffusivity (global N = 1; N = 37; Atlas Tract; Supplementary Table 5–6), (6) RSFC (within network, N = 13; between network, N = 78; Supplementary Table 7). Brain phenotypes were derived using Freesurfer segmentation (subcortical volumes; Dale et al. 1999), the Desikan-Killianry atlas (cortical thickness and surface area; (Desikan et al. 2006), Atlas Tract (FA, MD; Basser et al. 1994), and Gordon networks RSFC; Gordon et al. 2016). No task-related functional magnetic resonance imaging (fMRI) data were examined due to test–retest reliability concerns surrounding this method (Elliott et al. 2020).
AD genetic risk
Genetic data and quality control
Saliva samples were genotyped on the Smokescreen array (Baurley et al. 2016) by the Rutgers University Cell and DNA Repository (now incorporated with other companies as Sampled; https://sampled.com/). Genotyped calls were aligned to GRC37 (hg19). Rapid Imputation and COmputational PIpeLIne for Genome-Wide Association Studies (RICOPILI) (Lam et al. 2020) was used to perform quality control (QC) on the 11,099 individuals with available ABCD Study phase 3.0 genotypic data, using RICOPILI’s default parameters. The 10,585 individuals who passed QC checks were matched to broad self-reported racial groups using the ABCD Study parent survey. Of the 6787 parents/caregivers indicating that their child’s race was only “white,” 5561 of those individuals did not endorse any Hispanic ethnicity/origin. After performing a second round of genetic data QC on these sub-samples, 5556 non-Hispanic White individuals were retained in the analyses. Principal component analysis (PCA) in RICOPILI was used to confirm the genetic ancestry of these individuals by mapping onto the 1000 Genomes reference panel, resulting in a PCA-selected European-ancestry subset. The TOPMed imputation reference panel was used for imputation (Taliun et al. 2021). Imputation dosages were converted to best-guess hard-called genotypes, and only SNPs with Rsq > 0.8 and MAF > 0.01 were kept for PRS analyses.
Generating polygenic risk scores and APOE genotype
Genetic risk for AD was represented using 4 indices: (1) polygenic risk across the genome (ADPRS), (2) APOE rs429358 genotype (APOE), (3) polygenic risk for AD excluding the APOE region (ADPRS-APOE), and (4) a moderation analysis in which polygenic risk for AD excluding the APOE region was moderated by APOE rs429358 genotype (ADPRS-APOE x APOE genotype).
PRS-CS (Ge et al. 2019) was used to generate AD polygenic risk scores using effect size estimates from the largest GWAS of AD (N = 1,126,563; Wightman et al. 2021). Given the European ancestry background of the AD GWAS and the ABCD analytic sample, the corresponding European ancestry LD reference panel from the 1000 Genomes Project Phase 3 samples (available for download from https://github.com/getian107/PRScs) was used. The ‘auto’ function of PRS-CS was applied, allowing the software to learn the global shrinkage parameter from the data with 10,000 iterations and 5,000 burn-in. After deriving SNP weights using PRS-CS, PLINK 1.9’s—score command was used to produce PRS in the ABCD sample. For the creation of the PRS for AD excluding the APOE region (ADPRS-APOE); chr19:45,116,911–46,318,605). This definition was selected based on previous papers’ definitions of the APOE genomic region (e.g., Kunkle et al. 2019) and based on a regional association plot of GWAS signals around the APOE gene. APOE genotype was derived from the ABCD sample by using Plink 1.9’s—recode flag to generate a count of the number of ‘C’ “risk” alleles for the rs429358 SNP, which has been used to index APOE genetic risk (Cruchaga et al. 2012). Risk (i.e., “C”) allele counts for APOE rs429358 are given in Table 1.
Statistical analyses
Numeric data were scaled to a mean of 0 and standard deviation of 1 prior to analysis. PheWAS associations between genetic risk for AD (i.e., ADPRS, APOE, ADPRS-APOE, ADPRS-APOEx APOE) and phenotypes were estimated using independent mixed effects models in the lme4 R software package (Bates et al. 2015); the lmer() function was used for continuous outcomes (Bates et al. 2015), and the glmer() function was used for dichotomous outcomes (Austin 2010). All non-imaging models were nested by site and family ID while imaging models were nested by scanner and family ID to account for the non-independence of these data. Fixed effect covariates for all analyses included: the first 10 ancestry principal components, age, and sex (sex was removed for models where the outcome was a sex-specific phenotype, e.g. “Have you noticed a deepening of your voice?” (pds_m4_y) from the ABCD Youth Pubertal Development Scale and Menstrual Cycle Survey History). For brain structure regional estimates, a global index was also included as a covariate (Supplementary Table 6; i.e., total cortical thickness for regional cortical thickness analyses; total cortical and subcortical volume for cortical and subcortical gray matter volume, respectively; total surface area for regional surface area analyses; and average fractional anisotropy or mean diffusivity across all fibers for all DTI analyses). To ensure seed stability within generalized linear mixed-effects models (GLMER), all models were run seven times using the base R set.seed() function for the following randomly generated seeds: 10, 18, 29, 42, 73, 96, and 168. The marginal R2 of fixed effects was calculated using the MuMIn package (Bartoń 2009). R2 was calculated by using the difference of the marginal R2 of the genetic indice and the marginal R2 without the genetic indice. Given that there is no true R2 with logistic models, to calculate the R2 of the GLMER models Nagelkerke pseudo R2 was used (Nagelkerke et al. 1991).
To adjust for multiple testing, false discovery rate (FDR) and a Bonferroni-corrected phenome-wide significance threshold were used separately for psychosocial and behavioral phenotypes (0.05/1269 = 0.000039 Bonferroni alpha level) and each respective imaging modality: subcortical volume (0.05/35 = 0.00143 Bonferroni alpha level); cortical volume, cortical thickness, and surface area for each (0.05/68 = 0.00074 Bonferroni alpha level); mean diffusivity and fractional anisotropy for each (0.05/37 = 0.00135 Bonferroni alpha level); and RSFC (0.05/91 = 0.00055 Bonferroni alpha level) within each of our 4 genetic risk indices (i.e., ADPRS, APOE, ADPRS-APOE, ADPRS-APOE × APOE).
Results
None of the AD genetic risk indices (i.e., ADPRS, APOE, ADPRS-APOE, ADPRS-APOE × APOE) were significantly associated with any non-imaging or imaging phenotypes after either Bonferroni or FDR multiple testing correction (non-imaging: all |B|s or ORs < 2.42; all p > 0.00118; all pfdrs > 0.23, Fig. 1, Supplementary Table 4; imaging: all |B|s < 0.03; all ps > 0.003; all pfdrs > 0.22, Supplementary Tables 5–7). A monte-carlo based simulation power analysis using SIMR (Green and MacLeod 2016) suggested our most significant effect only had 58% power.

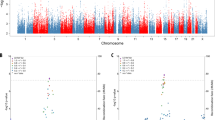
PheWAS Results for Non-imaging Phenotypes. Association between four genetic risk indices and cognitive, behavioral, and psychosocial phenotypes: A polygenic risk score (PRS) derived from the largest AD GWAS, B APOE rs429358 risk alleles, C a PRS that excludes the APOE4 region, and D the moderation of the PRS that excludes the APOE4 region by APOE4. Key for figure: 1a: ksads_10_324_p = “Symptom—Difficulty controlling worries Present,” ksads_10_322_p = “Symptom—Worry associated with defined symptom(s) Present,” devhx_19b_p = “At approximately what age (number of months) was he/she FIRST able to sit without assistance?,” and ksads_10_47_p = “Symptom—Worrying has lasted at least 6 months Present.” Key for Fig. 1B: asr_q07_p = “I brag”, devhx_19b_p = “At approximately what age (number of months) was he/she FIRST able to sit without assistance?,” parent_monitor_q4_y = “How often do you talk to your parent or guardian about your plans for the coming day, such as your plans about what will happen at school or what you are going to do with friends?,” and ksads_11_346_p = “Symptom—Impairment in functioning due to compulsions Past.” Key for Fig. 1C: medhx_6d = “Has he/she ever been to a doctor, a nurse, nurse practitioner, the emergency room or a clinic because Stitches,” cbcl_q11_p = “Clings to adults or too dependent,” asr_q14_p = “I cry a lot,” and devhx_14a3_p = “Did he/she have any of the following complications at birth? Blue at birth? Key for Fig. 1D: famhx_ss_matant1_prob_nrv_p = “maternal aunt 1 nerves/nervous breakdown problem,” kbi_p_c_spec_serv___7 = “Does your child receive special services at school?,” devhx_19b_p = “At approximately what age (number of months) was he/she FIRST able to sit without assistance?,” asr_q125_p = “In the past 6 months, on how many days were you drunk?,” and medhx_6i = “Has he/she ever been to a doctor, a nurse, nurse practitioner, the emergency room or a clinic because of a head injury.”
Psychosocial and behavioral phenotypes
No psychosocial or behavioral phenotypes, within any domain (i.e., cognition, screen time, demographics, substance, culture/environment, physical health, family mental health, and child mental health) were significantly associated with any index of AD genetic risk after either Bonferroni or FDR multiple testing correction. Across the four indices of genetic risk for AD: ADPRS, APOE, ADPRS-APOE, ADPRS-APOE × APOE genotype, six of the eight assessed domains had nominally significant associations (p < 0.01) and are reported in Tables 2, 3, 4. There were no nominally significant associations in either the Demographics or Screen Time domain. In total, across six domains (i.e., Child Mental Health, Family Mental Health, Physical Health/Development, Culture/Environment, Substances, Cognition) and ADPRS, APOE, and ADPRS-APOE, ADPRS-APOE × APOE genotype, there were 18, 12, 10, and 15 nominally significant associations, respectively (nominal significance considered as p < 0.01 due to the large number of phenotypes investigated). Specifically, the following number of nominally significant associations were observed for each AD genetic risk by domain: Child Mental Health (ADPRS = 10, APOE = 4, ADPRS-APOE = 3, ADPRS-APOE × APOE genotype = 7), Family Mental Health (ADPRS = 3, APOE = 4, ADPRS-APOE = 4, ADPRS-APOE × APOE genotype = 3), Physical Health/Development (ADPRS = 3, APOE = 3, ADPRS-APOE = 3, ADPRS-APOE × APOE genotype = 5), Culture/Environment (ADPRS = 0, APOE = 1, ADPRS-APOE = 0, ADPRS-APOE × APOE genotype = 0), Substance (ADPRS = 1, APOE = 0, ADPRS-APOE = 0, ADPRS-APOE × APOE genotype = 0), Cognition (ADPRS = 0, APOE = 1, ADPRS-APOE = 0, ADPRS-APOE × APOE genotype = 0. Below and reported in Tables 2, 3, 4, we briefly summarize the directionality of associations that were below an uncorrected p value threshold of 0.01 within each of these domains for ADPRS, APOE, and ADPRS-APOE. Nominally significant ADPRS-APOE × APOE interactions are not described below as no post-hoc tests were conducted to characterize the directionality of these moderation effects due to the lack of significant interactions when accounting for multiple testing.
Cognition
APOE risk alleles were associated with reduced performance on the Rey Auditory Verbal Learning Test, a neuropsychological assessment of auditory-verbal attention, memory, and learning (Total score: B = − 0.04, p = 0.0061, pfdr = 0.70). No other associations p < 0.01 were observed for any AD genetic risk index. No other cognition phenotypes were associated with any AD genetic index at even nominal p < 0.05 levels of significance.
Screen time
No associations ps < 0.01 were observed for any AD genetic risk index.
Demographics
No associations ps < 0.01 were observed for any AD genetic risk index.
Substances
Higher ADPRS was associated with greater substance accessibility (i.e., “If your child wanted to get a drug like cocaine, LSD, or amphetamines, how easy would it be for them to get some?”; B = 0.04, p = 0.008, pfdr = 0.64). No other associations ps < 0.01 were observed for any AD genetic risk index.
Culture/environment
APOE risk alleles were associated with talking more often to one’s parent/guardian about daily plans (B = 0.04, p = 0.001, pfdr = 0.43). No other associations ps < 0.01 were observed for any AD genetic risk index.
Physical health (inclusive of development)
Both higher ADPRS and APOE risk alleles were associated with a later age (in months) when being able to sit up by oneself as an infant/toddler (both Bs > 0.04, ps < 0.002, psfdr > 0.23) and being evaluated by a medical professional for a sprain (both Bs > 0.12, ps < 0.002, psfdr > 0.23). Higher ADPRS was associated with greater pubertal development among males (B = 0.05, p = 0.006, pfdr = 0.57) and APOE risk alleles were associated with more hospitalizations (B = 0.10, p = 0.005, pfdr = 0.66). Finally, ADPRS-APOE was associated with receiving stitches from a medical practitioner and birth complications as well as a (all |B|s > 0.08, all ps < 0.008, all psfdr > 0.77). No other associations < 0.01 were observed for any AD genetic risk index.
Family mental health
The caregivers (predominantly mothers) of individuals with high ADPRS reported increased bragging, being less mean, and increased intrusive thoughts (all |B|s > 0.02, all ps < 0.01, all psfdr > 0.23). Among those with more APOE risk alleles, caregivers reported more bragging and talking too much as well as being less mean to others and that their behavior is less changeable (all |B|s > 0.02, all ps < 0.01, all psfdr > 0.43). Caregivers of those with elevated ADPRS-APOE reported lower emotional disturbance (e.g., less anhedonia, not crying a lot), dependence on others, and difficulty making decision (all Bs < − 0.03, all ps < 0.01, all psfdr > 0.82). No other associations < 0.01 were observed for any AD genetic risk index.
Child mental health
Greater ADPRS was associated with reduced anxiety (e.g., difficulty controlling worries) and manic symptoms (e.g., racing thoughts) and impairment (e.g., clinically significant distress due to worry) as well as increased impairment due to compulsions (all |B|s > 0.14, all ps < 0.009, all psfdr > 0.23). APOE risk alleles were associated with greater compulsive symptoms (e.g., past compulsions) and impairment (e.g., past impairment in function due to compulsions) and reduced anxiety symptoms (i.e., difficulty controlling worries) (all |B|s > 0.17, all ps < 0.007, all psfdr > 0.43). Finally, greater ADPRS-APOE was associated with less clinging to adults/dependence and receipt of special services at school as well as greater sleep problems and insomnia (all |B|s > 0.04, all ps < 0.009, all psfdr > 0.77). No other associations p < 0.01 were observed for any AD genetic risk index. All corrected and uncorrected non-imaging phenotype results are in Supplementary Table 4.
Neuroimaging phenotypes
No brain phenotype, either global or regional, was significantly associated with any index of AD genetic risk, when adjusting for multiple testing using FDR or Bonferroni correction (all |B|s < 0.03; all ps > 0.003; all psfdr > 0.22; Supplementary Tables 5–7). The association between ADPRS-APOE and increased cerebellum volume and white matter approached significance with FDR correction (volume: right & left: both |B|s > 0.26, both ps < 0.007, both psfdr = 0.074; white matter volume: |B|= 0.01, p = 0.0058, pfdr = 0.074). The hippocampus, which was been previously associated with AD genetic risk in healthy samples, was not associated with any index of AD genetic risk (all |B|s < 0.02; all ps > 0.14, all psfdr > 0.68; Supplementary Table 5–7). Across the four indices of genetic risk for AD: ADPRS, APOE, ADPRS-APOE, ADPRS-APOE x APOE genotype, all eight imaging domains had nominally significant associations (P < 0.05) and are reported in Tables 2, 3, 4. In total, across the eight imaging domains (i.e., Whole Brain, Regional Subcortical Volume, Regional Cortical Thickness, Cortical Surface Area, Regional Mean Diffusivity, Regional Fractional Anisotropy, and RSFC) the four indices of genetic risk for AD (ADPRS, APOE, and ADPRS-APOE, ADPRS-APOE × APOE genotype) had 16, 20, 27, and 25 nominally significant associations, respectively. Specifically, the following number of nominally significant associations were observed for each AD genetic risk by imaging modality: Whole Brain (ADPRS = 0, APOE = 0, ADPRS-APOE = 3, ADPRS-APOE × APOE genotype = 0) Regional Subcortical Volume (ADPRS = 3, APOE = 4, ADPRS-APOE = 5, ADPRS-APOE × APOE genotype = 1), Regional Cortical Volume (ADPRS = 4, APOE = 2 ADPRS-APOE = 4, ADPRS-APOE × APOE genotype = 4), Regional Cortical Thickness (ADPRS = 3, APOE = 4, ADPRS-APOE = 5, ADPRS-APOE × APOE genotype = 4), Cortical Surface Area (ADPRS = 3, APOE = 3, ADPRS-APOE = 4, ADPRS-APOE × APOE genotype = 7), Regional Mean Diffusivity (ADPRS = 1, APOE = 3, ADPRS-APOE = 2, ADPRS-APOE × APOE genotype = 2), Regional Fractional Anisotropy (ADPRS = 0, APOE = 0, ADPRS-APOE = 1, ADPRS-APOE × APOE genotype = 2), and RSFC (ADPRS = 2, APOE = 4, ADPRS-APOE = 3, ADPRS-APOE × APOE genotype = 5). Below, and reported in Tables 2, 3, 4, we briefly summarize associations that were nominally significant, i.e., p < 0.05 uncorrected for multiple testing for ADPRS, APOE risk alleles, ADPRS-APOE. The ADPRS-APOE × APOE interactions are not described here, as post-hoc tests to characterize these interactions were not conducted due to the lack of significance when accounting for multiple testing.
Whole brain
ADPRS-APOE was associated with greater total, right, and left cortical volumes at nominal levels of significance (all |B|s > 0.023, all ps < 0.04, all pfdr = 0.22). No other nominally significant associations were observed for any AD genetic risk index. All whole brain results are in Supplementary Table 6.
Regional
Volume
Subcortical
Overall, AD genetic risk (i.e., ADPRS, APOE risk alleles, ADPRS-APOE) was associated with greater left and right Nucleus Accumbens volumes at nominal levels of significance (all |B|s > 0.02; all ps < 0.05, all psfdr > 0.10). ADPRS was also associated with increased right putamen volume, as well as decreased right thalamic volume (all |B|s > 0.01; all ps < 0.05, all psfdr > 0.18). APOE genotype was associated with decreased brain stem volume, cerebellar cortical volume, and right thalamic volume (all |B|s > 0.01; all ps < 0.04, all psfdr > 0.20). Finally, ADPRS-APOE was associated with increased cerebellar cortex and cerebellar white matter volume (all |B|s > 0.02; all ps < 0.02, all psfdr > 0.07).
Cortical
ADPRS was associated with increased volume in the right precentral gyrus, left superior temporal sulcus, and right lingual gyrus, as well as decreased volume in the right medial orbitofrontal cortex (all |B|s > 0.019; all ps < 0.05, all psfdr > 0.48). APOE risk alleles were associated with increased volume in the right paracentral lobule and left superior temporal sulcus (all |B|s > 0.029; all ps < 0.02, all psfdr = 0.52). Finally, ADPRS-APOE was associated with increased volume in the left precuneus, as well as decreased left caudal anterior cingulate, right inferior parietal, and left parahippocampal cortical volumes (all |B|s > 0.02; all ps < 0.03, all psfdr = 0.10). All regional Volume results are in Supplementary Table 5.
Cortical thickness
ADPRS was associated with decreased thickness in the left and right supramarginal regions as well as the right inferior temporal region and increased thickness in the left superior temporal sulcus region (all |B|s > 0.019, all ps < 0.04, all psfdr > 0.30). APOE risk alleles were associated with increased thickness in the both hemispheres of the cuneus and the left superior temporal sulcus, as well as decreased inferior temporal thickness (all |B|s > 0.02, all ps < 0.03, all psfdr > 0.13). ADPRS-APOE4 was associated with increased left middle temporal and right isthmus cingulate thickness, as well as decreased left and right parahippocampal and right precuneus thickness (all |B|s > 0.018, all ps < 0.05, all psfdr > 0.42). All regional Cortical Thickness results are in Supplementary Table 5.
Cortical surface area
ADPRS was associated with increased right paracentral and right inferior temporal surface area, as well as decreased left caudal middle frontal surface area (all |B|s > 0.018, all ps < 0.04, all psfdr = 0.73). APOE risk alleles were associated with increased surface area in the right paracentral and left superior temporal sulcus, as well as decreased left rostral anterior cingulate surface area (all |B|s > 0.02, all ps < 0.04, all psfdr = 0.81). ADPRS-APOE was associated with increased left precuneus and right inferior temporal surface area, as well as decreased left caudal anterior cingulate and right inferior parietal surface area (all |B|s > 0.019, all ps < 0.03, all psfdr > 0.19). All Cortical Surface Area results are in Supplementary Table 5.
Mean diffusivity
ADPRS was associated with increased mean diffusivity (MD) within the right anterior thalamic radiations (B = 0.02, p = 0.006, psfdr = 0.23). APOE risk alleles were associated with increased MD within the right anterior thalamic radiations and the left striatal inferior frontal cortex, as well as decreased MD within the right parahippocampal cingulum (all |B|s > 0.02, all ps < 0.03, all psfdr = 0.27). ADPRS-APOE4 was associated with increased MD within the left parietal superior longitudinal fasciculus and the right inferior-fronto-occipital fasciculus (B > 0.01, all ps < 0.05, all psfdr = 0.39). All regional Mean Diffusivity results are in Supplementary Table 5.
Fractional anisotropy
ADPRS-APOE was associated with decreased FA within the right inferior-fronto-occipital fasciculus (B = − 0.01, p = 0.037, psfdr = 0.61). No other nominally significant associations were observed for any AD genetic risk index. All regional Fractional Anisotropy results are in Supplementary Table 5.
Resting state functional connectivity
ADPRS was associated with less functional coupling within the cingulo-opercular network and between the salience network and ventral attention network (B < − 0.02, all ps < 0.05, all psfdr = 0.95). APOE risk alleles were associated with increased correlated activity between the retrosplenial temporal network and both the “none” network and the default network and negative correlations between the retrosplenial temporal network and both the visual network and the dorsal attention network (all |B|s > 0.03, all ps < 0.02, all psfdr = 0.97). ADPRS-APOE4 was associated with increased correlation between the dorsal attention network and sensorimotor hand network, between the fronto-parietal network and salience network, and between the auditory network and cingulo-parietal network (B > 0.02, all ps < 0.04, all psfdr > 0.33). All RSFC results are in Supplementary Table 7.
Discussion
We conducted a PheWAS of behavioral, psychosocial, and neuroimaging phenotypes in relation to four indices of genetic risk for AD (i.e., ADPRS, APOE, ADPRS-APOE, ADPRS-APOE × APOE) within individuals of genomically-defined European ancestry in the ABCD Study (Ns = 120–5556). No phenotypes (N = 1687) were associated with any index of the four AD genetic risk indices after correction for multiple testing using Bonferroni or FDR. All nominally significant observed effects were small (Ranges: |B|s = 0.087–0.71; Ors 0.660–1.3). These null associations contrast positive associations observed between polygenic risk scores observed in the ABCD Study sample. Notably, as other psychiatric PRS (e.g., schizophrenia, cannabis use disorder, depression, etc.) have been associated with behavioral, environmental, and neuroimaging phenotypes within the ABCD Study sample (Johnson et al. 2020; Paul et al. 2021; Ohi et al. 2021; Karcher et al. 2022; Joo et al. 2022), our null findings cannot be attributed to broader poor polygenic prediction within this sample. Indeed, our observed null results raise the possibility that genetic risk for AD may not be phenotypically expressed during middle childhood or that any associations would be characterized by small effect sizes that our study (N = 5556) was underpowered to detect. In light of prior evidence linking AD genetic risk in healthy adults to cognition and brain structure (Bellenguez et al. 2022), it remains possible that individual differences in these phenotypes linked to genetic risk for AD do not emerge until adolescence and/or young adulthood when neurodevelopment transitions from growth to pruning (Tiemeier et al. 2010; Ladouceur et al. 2019; Sakai 2020).
Null and nominally significant associations
A few null associations and patterns of nominally significant association warrant discussion. First, contrary to hypotheses and prior studies in healthy adults (Fleisher et al. 2005; O’Dwyer et al. 2012; Evans et al. 2020; Walhovd et al. 2020; Murray et al. 2021), AD genetic risk was not associated with cognition or hippocampal volume in middle childhood. While APOE risk alleles were nominally associated with reduced auditory-verbal attention/memory/learning (Rey Auditory Verbal Learning Test Total Scores), this association did not approach significance when adjusting for multiple testing using our least stringent adjustment (FDR), and no other cognition phenotypes were associated with any index of genetic risk at nominal levels of significance. It is unlikely that this reflects a false negative association resulting from PheWAS multiple testing burden, as this association remains non-significant when only implementing multiple testing correction for cognition and hippocampal volumes across indices of AD genetic risk (N = 64 total tests; pfdr > 0.38). When interpreted alongside evidence from adult studies linking AD genetic risk to reduced cognition and hippocampal volume (Ns = 44–2690; (Fleisher et al. 2005; O’Dwyer et al. 2012; Evans et al. 2020; Walhovd et al. 2020; Murray et al. 2021), our null findings raise the possibility that lower cognition and smaller hippocampal volumes observed in healthy adults at elevated genetic risk for AD may arise after middle childhood.
Second, increased cerebellum volume and white matter among individuals with higher AD ADPRS-APOE scores approached significance with FDR adjustment for multiple testing correction within this modality. Notably, this observed effect was small (|B|= 0.0286 [95% CI 0.0097–0.0478]) and does approach significance if adjusting for all phenotypes examined simultaneously in the subcortical volume modality Pfdr = 0.069) and does not approach significance when applying FDR to all imaging phenotypes (Pfdr = 0.283). Reports of both larger (Lin et al. 2020) and smaller cerebellum (particularly gray matter; Gellersen et al. 2021) volume have been reported in AD dementia and related phenotypes (e.g., MCI; Jacobs et al. 2018). The effect size we observed (|B|= 0.0286, R2 = 0.0009) was drastically smaller than these phenotypic associations observed in adult cases (|B|= 0.05; Lin et al. 2020, Cohen’s D < 0.1; Gellersen et al. 2021). Other nominally significant brain structure findings (e.g., greater cortical volumes associated with ADPRS-APOE and increased Nucleus Accumbens volumes associated with all AD genetic risk indices) run counter to some observations in AD (Nie et al. 2017) as well as adults at genetic risk (Muir et al. 2021). Overall, there was a general pattern of nominally significant associations between AD risk and larger brain volumes. It is important to consider this pattern of findings in the context of the age of the sample (i.e., middle childhood). During typical development, there is extensive neural growth during middle childhood, after which a period of extensive pruning begins in adolescence, which slows in adulthood and then accelerates again in later life (Tiemeier et al. 2010; Ladouceur et al. 2019; Sakai 2020). While it is plausible that AD genetic risk may manifest as potentiated trajectories across development (i.e., including growth during childhood and pruning that begins in adolescence), the present pattern of nominally significant associations would need to be observed in independent samples before credence could be given to this possibility.
Third, nominally significant associations between AD genetic risk and development were observed. In particular both ADPRS and APOE genotype were nominally associated with delayed infant development (i.e., a later age when an infant is first able to sit up by themself) and greater pubertal development among males. This directional association may be partially explained by the prevailing theory that later infant motor development may be related to worse cognitive and motor functioning that has been previously associated with cognitive ability (Murray et al. 2007). Earlier pubertal timing is also associated with worse cognitive functioning (Ghassabian et al. 2016). Thus, this may potentially reflect broad developmental signs associated with poor cognitive outcomes that are shared with AD genetic risk.
Fourth, several nominally significant associations were observed between different indices of AD genetic risk and increased injuries by middle childhood (e.g., more hospitalizations; evaluation by medication professionals for a sprain, receiving stitches from a medical practitioner, and birth complications). There is a wide variety of evidence suggesting that mild traumatic brain injury is associated with the future development of AD (Graham et al. 2022). It is possible that AD genetic risk may emerge through gene-environment correlation (e.g., increasing the likelihood of injury; Graham et al. 2022) and/or that AD genetic risk may moderate recovery following injury (e.g., inflammation) that increases AD risks (Alexander et al. 2007). However, there also was evidence of opposing associations (i.e., reduced broken bones) that should generate caution in overly interpreting this pattern of association with injury without additional evidence.
Fifth, there was nominally significant evidence that AD genetic risk was associated with reduced psychopathology symptoms (e.g., anxiety, anhedonia, anxiety) in children and their caregivers (predominantly mothers) and less receipt of special services at school, but increased compulsive behavior in children. As anxiety, psychopathology, and related behavior have been associated with increased risk for AD and related risk factors (e.g., mild cognitive impairment; Stafford et al. 2022), these findings counter what has been observed in adults, with the exception of compulsions which have been linked to AD risk (Dondu et al. 2015).
Overall, while there was little consistency in nominally significant associations across the three genetic indices of AD risk (ADPRS, APOE, ADPRS-APOE), the following phenotypes emerged in both the ADPRS and APOE analyses: Child Mental Health (ksads_10_324_p, “Symptom—Difficulty controlling worries Present” and ksads_11_346_p, “Symptom—Impairment in functioning due to compulsions Past”), Family Mental Health (asr_q07_p “I brag”), and Physical Health/Development: (medhx_6b, “Has he/she ever been to a doctor, a nurse, nurse practitioner, the emergency room or a clinic because of sprains”). This pattern may partially arise through the large risk conferred by APOE genotypes that heavily weights PRS; this interpretation is further supported by the lack of overlap between ADPRS and APOE with ADPRS-APOE. Interestingly, only the ADPRS had nominally significant association in the Substance domain (su_risk_p_5, “If your child wanted to get a drug like cocaine, LSD, or amphetamines, how easy would it be for them to get some?”), and only the APOE genotype had nominally significant results in Cognition (RAVLT, “Rey Auditory Verbal Learning Test total score.”
Limitations
The large sample size and deeply phenotyped sample permitted the PheWAS approach; however, the revealed null associations should also be interpreted in the context of study limitations. First, this is a cross-sectional study in a European ancestry subsample of individuals volunteering for research. This limits study generalizability and prohibits the evaluation of change, which may be especially important for neurodegenerative disorders. Second, the current PheWAS only estimated the associations between indices of genetic risk for late-onset AD, which has a heritability rate of 70–80% (Bekris et al. 2010). Importantly, non-mendelian early onset AD (< 65 years old) has a heritability rate of 92–100%, but there currently is not sufficient GWAS data from either national or international representative samples due to its low incidence and prevalence rate, (Reitz et al. 2020). Third, it is possible that our PheWAS did not include important indicators of AD risk that may be present during childhood. PheWAS variables were constrained by those measured in the ABCD study and do not include indices that may be more proximal to AD (e.g., to the best of the author’s knowledge, the ABCD study does not collect family history of AD, dementia, or assess parental cognition) and others that have been previously linked to AD risk (e.g., inflammation; Zhang et al. 2022).
Fourth, the discovery GWAS of AD (Wightman et al. 2021) used to generate the AD PRS applied case–control association mapping by proxy, a method which results in potentially less specific accuracy of identifying AD, compared to the case–control approach where a clinical or pathological diagnosis of AD is used (Liu et al. 2017). A recent report highlights low SNP heritability (0.03), which may limit associations with the polygenic scores (Escott-Price and Hardy 2022). However, the case–control approach can be difficult to conduct, as collecting a large enough sample size can be both time consuming and expensive (Liu et al. 2017). Thus, the tradeoff for using the case–control association mapping by proxy method which generates “noisier” data is potentially mitigated by this increased sample size, which in turn reduces false positive associations as well as improves discovery rate (Hong and Park 2012) of significant loci of AD risk. It will be important to continue to examine this association as GWASs with more cases become available (Bellenguez et al. 2022) and evaluate the impact of proxy case inclusion on observed estimates.
Conclusions
Associations between genetic risk for AD and the psychosocial and neural phenotypes in childhood that we examined are small in magnitude. We did not find evidence for significant associations between AD Genetic risk with cognitive, behavioral, psychosocial, or imaging phenotypes in the European ancestry sample of ABCD during middle childhood. These data suggest that the manifestation of genetic risk for AD may not emerge, at least measurably, until adolescence and/or young adulthood.
Data availability
All ABCD data used in this study are available through the National Institute of Mental Health Data Archive (NDA), which may be accessed here: https://nda.nih.gov/.
Code availability
References
Albert MS, DeKosky ST, Dickson D et al (2011) The diagnosis of mild cognitive impairment due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement 7:270–279. https://doi.org/10.1016/j.jalz.2011.03.008
Alexander S, Kerr ME, Kim Y et al (2007) Apolipoprotein E4 allele presence and functional outcome after severe traumatic brain injury. J Neurotrauma 24:790–797. https://doi.org/10.1089/neu.2006.0133
Austin PC (2010) Estimating multilevel logistic regression models when the number of clusters is low: a comparison of different statistical software procedures. Int J Biostat 6:16. https://doi.org/10.2202/1557-4679.1195
Bartoń K (2009) MuMIn: multi-model inference
Basser PJ, Mattiello J, Lebihan D (1994) Estimation of the effective self-diffusion tensor from the NMR spin echo. J Magn Reson Ser B 103:247–254. https://doi.org/10.1006/jmrb.1994.1037
Bates D, Mächler M, Bolker B, Walker S (2015) Fitting linear mixed-effects models using lme4. J Stat Softw 67:1–48. https://doi.org/10.18637/jss.v067.i01
Baurley JW, Edlund CK, Pardamean CI et al (2016) Smokescreen: a targeted genotyping array for addiction research. BMC Genomics 17:145. https://doi.org/10.1186/s12864-016-2495-7
Bekris LM, Yu C-E, Bird TD, Tsuang DW (2010) Genetics of Alzheimer disease. J Geriatr Psychiatry Neurol 23:213–227. https://doi.org/10.1177/0891988710383571
Bellenguez C, Küçükali F, Jansen IE et al (2022) New insights into the genetic etiology of Alzheimer’s disease and related dementias. Nat Genet 54:412–436. https://doi.org/10.1038/s41588-022-01024-z
Cruchaga C, Kauwe JSK, Nowotny P et al (2012) Cerebrospinal fluid APOE levels: an endophenotype for genetic studies for Alzheimer’s disease. Hum Mol Genet 21:4558–4571. https://doi.org/10.1093/hmg/dds296
Dale AM, Fischl B, Sereno MI (1999) Cortical surface-based analysis I. Segmentation and surface reconstruction. Neuroimage 9:179–194. https://doi.org/10.1006/nimg.1998.0395
Dean DC, Jerskey BA, Chen K et al (2014) Brain differences in infants at differential genetic risk for late-onset Alzheimer disease: a cross-sectional imaging study. JAMA Neurol 71:11–22. https://doi.org/10.1001/jamaneurol.2013.4544
Desikan RS, Ségonne F, Fischl B et al (2006) An automated labeling system for subdividing the human cerebral cortex on MRI scans into gyral based regions of interest. Neuroimage 31:968–980. https://doi.org/10.1016/j.neuroimage.2006.01.021
Dondu A, Sevincoka L, Akyol A, Tataroglu C (2015) Is obsessive-compulsive symptomatology a risk factor for Alzheimer-type dementia? Psychiatry Res 225:381–386. https://doi.org/10.1016/j.psychres.2014.12.010
Elliott ML, Knodt AR, Ireland D et al (2020) What is the test-retest reliability of common task-functional MRI measures? New empirical evidence and a meta-analysis. Psychol Sci 31:792–806. https://doi.org/10.1177/0956797620916786
Escott-Price V, Hardy J (2022) Genome-wide association studies for Alzheimer’s disease: bigger is not always better. Brain Commun 4:fcac125. https://doi.org/10.1093/braincomms/fcac125
Evans SL, Dowell NG, Prowse F et al (2020) Mid age APOE ε4 carriers show memory-related functional differences and disrupted structure-function relationships in hippocampal regions. Sci Rep 10:3110. https://doi.org/10.1038/s41598-020-59272-0
Filippini N, MacIntosh BJ, Hough MG et al (2009) Distinct patterns of brain activity in young carriers of the APOE-epsilon4 allele. Proc Natl Acad Sci USA 106:7209–7214. https://doi.org/10.1073/pnas.0811879106
Fleisher A, Grundman M, Jack CR Jr et al (2005) Sex, apolipoprotein E ε4 status, and hippocampal volume in mild cognitive impairment. Arch Neurol 62:953–957. https://doi.org/10.1001/archneur.62.6.953
GBD 2019 Dementia Forecasting Collaborators (2022) Estimation of the global prevalence of dementia in 2019 and forecasted prevalence in 2050: an analysis for the Global Burden of Disease Study 2019. Lancet Public Health 7:e105–e125.https://doi.org/10.1016/S2468-2667(21)00249-8
Ge T, Chen C-Y, Ni Y et al (2019) Polygenic prediction via Bayesian regression and continuous shrinkage priors. Nat Commun 10:1776. https://doi.org/10.1038/s41467-019-09718-5
Gellersen HM, Guell X, Sami S (2021) Differential vulnerability of the cerebellum in healthy ageing and Alzheimer’s disease. NeuroImage 30:102605. https://doi.org/10.1016/j.nicl.2021.102605
Ghassabian A, Sundaram R, Bell E et al (2016) Gross motor milestones and subsequent development. Pediatrics 138:e20154372. https://doi.org/10.1542/peds.2015-4372
Gordon EM, Laumann TO, Adeyemo B et al (2016) Generation and evaluation of a cortical area parcellation from resting-state correlations. Cereb Cortex 26:288–303. https://doi.org/10.1093/cercor/bhu239
Grabher BJ (2018) Effects of Alzheimer disease on patients and their family. J Nucl Med Technol 46:335–340. https://doi.org/10.2967/jnmt.118.218057
Graham A, Livingston G, Purnell L, Huntley J (2022) Mild traumatic brain injuries and future risk of developing Alzheimer’s disease: systematic review and meta-analysis. J Alzheimers Dis 87:969–979. https://doi.org/10.3233/JAD-220069
Green P, MacLeod CJ (2016) SIMR: an R package for power analysis of generalized linear mixed models by simulation. Methods Ecol Evol 7:493–498. https://doi.org/10.1111/2041-210X.12504
Hendriks S, Peetoom K, Bakker C et al (2021) Global prevalence of young-onset dementia: a systematic review and meta-analysis. JAMA Neurol 78:1080–1090. https://doi.org/10.1001/jamaneurol.2021.2161
Hong EP, Park JW (2012) Sample size and statistical power calculation in genetic association studies. Genomics Inf 10:117–122. https://doi.org/10.5808/GI.2012.10.2.117
Jacobs HIL, Hopkins DA, Mayrhofer HC et al (2018) The cerebellum in Alzheimer’s disease: evaluating its role in cognitive decline. Brain J Neurol 141:37–47. https://doi.org/10.1093/brain/awx194
Johnson EC, Demontis D, Thorgeirsson TE et al (2020) A large-scale genome-wide association study meta-analysis of cannabis use disorder. Lancet Psychiatry 7:1032–1045. https://doi.org/10.1016/S2215-0366(20)30339-4
Joo YY, Moon S-Y, Wang H-H et al (2022) Association of genome-wide polygenic scores for multiple psychiatric and common traits in preadolescent youths at risk of suicide. JAMA Network Open 5:e2148585. https://doi.org/10.1001/jamanetworkopen.2021.48585
Korologou-Linden R, Bhatta L, Brumpton BM et al (2022) The causes and consequences of Alzheimer’s disease: phenome-wide evidence from Mendelian randomization. Nat Commun 13:4726. https://doi.org/10.1038/s41467-022-32183-6
Karcher NR, Paul SE, Johnson EC et al (2022) Psychotic-like experiences and polygenic liability in the adolescent brain cognitive development study. Biol Psychiatry Cognit Neurosci Neuroimaging 7:45–55. https://doi.org/10.1016/j.bpsc.2021.06.012
Kunkle BW, Grenier-Boley B, Sims R et al (2019) Genetic meta-analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates Aβ, tau, immunity and lipid processing. Nat Genet 51:414–430. https://doi.org/10.1038/s41588-019-0358-2
Kunkle BW, Schmidt M, Klein H-U et al (2021) Novel Alzheimer disease risk loci and pathways in African American individuals using the African genome resources panel: a meta-analysis. JAMA Neurol 78:102–113. https://doi.org/10.1001/jamaneurol.2020.3536
Ladouceur CD, Kerestes R, Schlund MW et al (2019) Neural systems underlying reward cue processing in early adolescence: the role of puberty and pubertal hormones. Psychoneuroendocrinology 102:281–291. https://doi.org/10.1016/j.psyneuen.2018.12.016
Lam M, Awasthi S, Watson HJ et al (2020) RICOPILI: rapid imputation for COnsortias PIpeLIne. Bioinformatics 36:930–933. https://doi.org/10.1093/bioinformatics/btz633
Liu JZ, Erlich Y, Pickrell JK (2017) Case-control association mapping by proxy using family history of disease. Nat Genet 49:325–331. https://doi.org/10.1038/ng.3766
Livingston G, Huntley J, Sommerlad A et al (2020) Dementia prevention, intervention, and care: 2020 report of the Lancet Commission. Lancet 396:413–446. https://doi.org/10.1016/S0140-6736(20)30367-6
Martin AR, Kanai M, Kamatani Y et al (2019) Clinical use of current polygenic risk scores may exacerbate health disparities. Nat Genet 51:584–591. https://doi.org/10.1038/s41588-019-0379-x
Mol MO, van der Lee SJ, Hulsman M et al (2022) Mapping the genetic landscape of early-onset Alzheimer’s disease in a cohort of 36 families. Alzheimers Res Ther 14:77. https://doi.org/10.1186/s13195-022-01018-3
Muir AM, Ching C, Santhalingam V et al (2021) The relationship between APOE genotype and subcortical volume: a UK Biobank study (N=36,920). Alzheimer’s Dementia 17:e055650. https://doi.org/10.1002/alz.055650
Murray GK, Jones PB, Kuh D, Richards M (2007) Infant developmental milestones and subsequent cognitive function. Ann Neurol 62:128–136. https://doi.org/10.1002/ana.21120
Murray AN, Chandler HL, Lancaster TM (2021) Multimodal hippocampal and amygdala subfield volumetry in polygenic risk for Alzheimer’s disease. Neurobiol Aging 98:33–41. https://doi.org/10.1016/j.neurobiolaging.2020.08.022
Nagelkerke NJD Miscellanea A note on a general definition of the coefficient of determination
Nichols E, Szoeke CEI, Vollset SE et al (2019) Global, regional, and national burden of Alzheimer’s disease and other dementias, 1990–2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol 18:88–106. https://doi.org/10.1016/S1474-4422(18)30403-4
Nie X, Sun Y, Wan S et al (2017) Subregional structural alterations in hippocampus and nucleus accumbens correlate with the clinical impairment in patients with Alzheimer’s disease clinical spectrum: parallel combining volume and vertex-based approach. Front Neurol 8:399. https://doi.org/10.3389/fneur.2017.00399
O’Dwyer L, Lamberton F, Matura S et al (2012) Reduced hippocampal volume in healthy young ApoE4 carriers: an MRI study. PLoS ONE 7:e48895. https://doi.org/10.1371/journal.pone.0048895
Ohi K, Ochi R, Noda Y et al (2021) Polygenic risk scores for major psychiatric and neurodevelopmental disorders contribute to sleep disturbance in childhood: Adolescent Brain Cognitive Development (ABCD) Study. Transl Psychiatry 11:1–11. https://doi.org/10.1038/s41398-021-01308-8
Paul SE, Hatoum AS, Fine JD et al (2021) Associations between prenatal cannabis exposure and childhood outcomes: results from the ABCD study. JAMA Psychiatry 78:64–76. https://doi.org/10.1001/jamapsychiatry.2020.2902
Reitz C, Rogaeva E, Beecham GW (2020) Late-onset vs nonmendelian early-onset Alzheimer disease: a distinction without a difference? Neurol Genet 6:e512. https://doi.org/10.1212/NXG.0000000000000512
Sakai J (2020) Core Concept: How synaptic pruning shapes neural wiring during development and possibly, in disease. Proc Natl Acad Sci USA 117:16096–16099. https://doi.org/10.1073/pnas.2010281117
Sims R, Hill M, Williams J (2020) The multiplex model of the genetics of Alzheimer’s disease. Nat Neurosci 23:311–322. https://doi.org/10.1038/s41593-020-0599-5
Stafford J, Chung WT, Sommerlad A et al (2022) Psychiatric disorders and risk of subsequent dementia: systematic review and meta-analysis of longitudinal studies. Int J Geriatr Psychiatry 37:5711. https://doi.org/10.1002/gps.5711
Taliun D, Harris DN, Kessler MD et al (2021) Sequencing of 53,831 diverse genomes from the NHLBI TOPMed Program. Nature 590:290–299. https://doi.org/10.1038/s41586-021-03205-y
Tiemeier H, Lenroot RK, Greenstein DK et al (2010) Cerebellum development during childhood and adolescence: a longitudinal morphometric MRI study. Neuroimage 49:63–70. https://doi.org/10.1016/j.neuroimage.2009.08.016
Volkow ND, Koob GF, Croyle RT et al (2018) The conception of the ABCD study: from substance use to a broad NIH collaboration. Dev Cogn Neurosci 32:4–7. https://doi.org/10.1016/j.dcn.2017.10.002
Walhovd KB, Fjell AM, Sørensen Ø et al (2020) Genetic risk for Alzheimer disease predicts hippocampal volume through the human lifespan. Neurol Genet 6:e506. https://doi.org/10.1212/NXG.0000000000000506
Wang H, Abbas KM, Abbasifard M et al (2020) Global age-sex-specific fertility, mortality, healthy life expectancy (HALE), and population estimates in 204 countries and territories, 1950–2019: a comprehensive demographic analysis for the Global Burden of Disease Study 2019. The Lancet 396:1160–1203. https://doi.org/10.1016/S0140-6736(20)30977-6
Wightman DP, Jansen IE, Savage JE et al (2021) A genome-wide association study with 1,126,563 individuals identifies new risk loci for Alzheimer’s disease. Nat Genet 53:1276–1282. https://doi.org/10.1038/s41588-021-00921-z
Zhang Z, Wang M, Liu X (2022) C-reactive protein and risk of Alzheimer’s disease. Neurobiol Aging 109:259–263. https://doi.org/10.1016/j.neurobiolaging.2021.08.010
Acknowledgements
We are thankful to families who have participated in the ABCD Study as well as study staff and investigators. We thank Carlos Cruchaga for guidance on APOE genotype coding.
Funding
This study was funded by R01DA054750 (RB, AA). AJG was supported by NSF DGE-213989. SEP was supported by F31AA029934. NRK was supported by K23MH12179201. ECJ was supported by K01DA051759. ASH was supported by K01AA030083. Data for this study were provided by the Adolescent Brain Cognitive Development (ABCD) study, which was funded by the National Institutes of Health (grants U01DA041022, U01DA041025, U01DA041028, U01DA041048, U01DA041089, U01DA041093, U01DA041106, U01DA041117, U01DA041120, U01DA041134, U01DA041148, U01DA041156, U01DA041174, U24DA041123, and U24DA041147) and additional federal partners (https://abcdstudy.org/federal-partners.html).
Author information
Authors and Affiliations
Contributions
AJG, SEP, NRK, IN, LB, ISH cleaned the phenotypic data. AJG, SEP, ECJ, SC, ASH cleaned the genomic data and calculated/coded genetic risk (i.e., PRS, APOE4). AJG, SEP, ASH conducted analyses. AJG and RB conceptualized the study and AJG, RB, ASH, ECJ, NRK, and SEP drafted the initial manuscript. All coauthors provided input on study conceptualization and edited the manuscript with important intellectual content.
Corresponding author
Ethics declarations
Conflict of interest
Aaron J. Gorelik, Sarah E. Paul, Nicole R. Karcher, Emma C. Johnson, Isha Nagella, Lauren Blaydon, Hailey Modi, Isabella S. Hansen, Sarah M.C. Colbert, David A.A. Baranger, Sara A. Norton, Isaiah Spears, Brian Gordon, Wei Zhang, Patrick L. Hill, Thomas F. Oltmanns, Janine D. Bjisterbosch, Arpana Agrawal, Alexander S. Hatoum, and Ryan Bogdan declare that they have no conflict of interest.
Ethical approval
Working with ABCD NDA data was approved by the Washington University in St. Louis Institutional Review Board: IRB ID#201708123.
Human and Animal Rights and Informed consent
All data was obtained through the Adolescent Brain and Cognitive Development study. Informed consent was handled by each site. The ABCD data is publicly available (secondary data analysis). Informed consent was obtained from each site before data collection from the Childs parent or guardian. Data was deanonymized before download via the public NDA App. ABCD has extensive protocols for participant consent, safety, and anonymity, see https://doi.org/10.1016/j.dcn.2017.06.005.
Additional information
Handling Editor: Chun Chieh Fan.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Gorelik, A.J., Paul, S.E., Karcher, N.R. et al. A Phenome-Wide Association Study (PheWAS) of Late Onset Alzheimer Disease Genetic Risk in Children of European Ancestry at Middle Childhood: Results from the ABCD Study. Behav Genet 53, 249–264 (2023). https://doi.org/10.1007/s10519-023-10140-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10519-023-10140-3