
Abstract
This study explores power assumptions relating to extended pedigree designs (EPD) and classical twin designs (CTD). We conducted statistical analyses to compare the power of the two designs for examining neuroimaging phenotypes, varying heritability and varying whether shared environmental variance is fixed or free. Results indicated that CTDs have more power to estimate heritability, with the exception of one condition: in EPDs, the power increases relative to CTDs when shared environmental variance contributes to sibling similarity only. We additionally show that assuming a priori that shared environmental effects play no role in a phenotype—as is commonly done in pedigree designs—can lead to substantially biased heritability estimates. General results indicate that both CTDs and EPDs obtain quite precise heritability estimates. Finally, we discuss methodological considerations relating to assumptions about age effects and shared environment.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Almost 100 years after Fisher and Wright first conceptualized heritability, it remains central to the examination of phenotypic variability (Visscher et al. 2008). The biometric modeling approach to estimating heritability yields apparent consistency of estimates across classical twin designs (CTDs) and extended pedigree designs (EPDs). The CTD has traditionally been considered optimal for estimating heritability (Neale and Cardon, 1992). However, the EPD might be superior because it has: (1) greater potential for identification of quantitative trait loci (QTL); (2) may have less confounding of genetic and shared environmental effects; and (3) may have increased statistical power to detect heritability (Winkler et al. 2009; Glahn et al. 2010). It is true that the large pedigree design can be advantageous when it comes to locating QTL (Blangero et al. 2003; Dolan et al. 1999), but the other two points must be formally tested. Here we sought to formally test the second and third hypotheses by comparing results under different assumptions in both the CTD and ETD.
General methodological considerations
It has been established that extending CTDs by combining them with additional data from other family members increases the utility of the CTD (Neale and Cardon 1992; Posthuma and Boomsma 2000; Truett et al. 1994; Keller et al. 2009). For example, extended twin designs can help to differentiate the effects of shared environment versus assortative mating and can resolve confounds of gene-by-common environment interactions. EPDs that lack data from twins, however, require assumptions that may be questionable (Kendler and Neale 2009).
General heritability estimation
Heritability is the genetic variance divided by the total (genetic + environmental) variance; therefore, to calculate heritability, one must be able to separate the genetic and environmental variance components. In the CTD, variance components are usually separated into those that reflect additive genetic (A), common or shared environmental (C), and unique, individual-specific environmental (E) influences; typically referred to as an ACE model. The standardized additive genetic variance (A) is the heritability (Neale and Cardon 1992). It is also worth noting that issues regarding heritability estimates apply equally to the calculation of genetic correlations, going beyond the heritability of individual variables and estimate the correlations between the genetic factors influencing two different variables.
Assumption of reduced genetic and shared environmental confounds in pedigree designs
One assumption of the superiority of the EPD is that having multiple households within a pedigree reduces confounds of genetic influences with shared environmental influences. However, shared environment, proposed to result in similarity between relatives, cannot simply be equated with one’s household environment. As noted by Carey (2003), factors within a family household are part of the statistical shared environment only if they also make relatives similar. It is very possible for factors outside the household (e.g., school quality) to make relatives similar. Similarly, factors within the same household can make relatives different (e.g., differential treatment by parents, or the effects of parental behavior on children differing from the impact of children’s behavior on parents) (Coon and Carey 1989). In these cases, the extra-household factors are common environmental influences and the intra-household factors are unique environmental influences.
In actual practice, the EPD has almost always been implemented with statistical models that ignore C completely and include only the A and E variance components. Thus, most estimates of heritability in pedigree studies are based solely on AE models. In other words, in that analytic strategy it is presumed a priori that C is zero. This approach can be problematic if C is not close to zero.
An example from some neuroimaging data illustrates this point. In a univariate ACE model, heritability (A) for right rostral middle frontal gyrus surface area was estimated at .28 [95 % confidence interval (CI) .14; .59], shared environmental influences (C) were estimated at .29 (.00; .47), and unique environmental influences (E) were estimated at .43 (.34; .53). The 95 % CIs were very wide for A and C, and C was nonsignificant. In the AE model, in which C is dropped, the heritability estimate jumps to .57 with a much narrower 95 % CI (.46; .67).
Two key points should be readily apparent from this example. First, if one began with this AE model and ignored the C component of variance entirely, the result would give the illusion that the proportion of genetic variance is essentially double what we would consider to be the less biased estimate obtained from the ACE model. Because the E estimates were virtually unchanged, it can be seen that the shared environmental variance has essentially been added to the genetic variance. Dropping C resulted in a substantial narrowing of the confidence interval for A, which also means that A will more often be significant than it would in an ACE model. Second, we cannot know in advance what the shared environmental effects will be for any given phenotype in any particular sample or dataset. The heritability estimate changed substantially in our example, but if C were near zero the heritability estimate in the univariate analysis would, in most cases, change very little.
Therefore, it is not possible to know whether a particular heritability estimate is substantially biased without first considering the full ACE models. Given the limitations of using household as a proxy for common environmental influences and the frequent omission of the C component of variance in pedigree studies, we think it is more accurate to say that the EPD provide estimates of “familiality” rather than heritability (Kendler and Neale 2009). A method that enables one to separate shared environmental from genetic and unique environmental influences (e.g., the CTD or adoption study instead of nuclear families) is crucial for determining accurate estimates of heritability with confidence.
This issue is particularly important for those phenotypes about which the extent of shared environmental influences is relatively unknown (for example functional, as opposed to structural, neuroimaging phenotypes), and for those phenotypes that have been shown to have significant C effects (e.g., social attitudes, educational attainment, musical ability, substance use initiation) (Heath et al. 1985; Eaves et al. 1999; Heath et al. 1993; Kendler et al. 1999; Schork and Schork 1993; Williams and Blangero 1999). In those cases, heritability estimates based on modeling extended pedigree data without a C component may be seriously biased.
Assumption about the statistical power of twin versus pedigree designs
Other things being equal, statistical power to detect QTL increases as pedigree size increases (Glahn et al. 2010; Schork and Schork 1993; Williams and Blangero 1999). Consequently, the sibling-pair design has the lowest power to detect QTL among pedigree studies. It could be argued that the same logic applies to heritability estimates. However, this assertion ignores the fact that the nature of the genetic relationships and the amount of useful information are very different in twin and sibling-pair studies. Non-twin and dizygotic twin siblings share, on average, 50 % of their genes, but MZ twins share 100 % of their genes. Therefore, the CTD adds substantial power to determine heritability because it is unique in being able to include comparisons of 100 versus 50 % of shared genes.
Extended pedigrees include pairs of first-degree relatives who share 50 % of their genes, pairs of second-degree relatives who share 25 % of their genes, and so on. For example, first cousins (third-degree relatives) share approximately 12.5 % of their genes. Therefore, their maximum genetic similarity is r = .125. Second cousins (fourth-degree) relatives share approximately 6.25 % of their genes, and the difference between third- and fourth-degree relatives is only 6.25 %. Power to detect a significant difference between small correlations is limited. In this case, correlations for a 100 % additive genetic trait would be .125 and .0625. To have power to detect heritability of 50 %, one would need approximately 100 times as many participants as would be needed in a classical twin design, because the difference in allele sharing between the two types of pairs would be 50 % for twin versus 6.25 % for first versus second cousins.
Thus, with greatly extended pedigrees, the gain from having increased numbers of participants diminishes substantially as increasingly distant relatives are included. On the other hand, the number of effective pairs does increase with larger pedigrees, so ideally one would pursue pedigrees with, for example, large sibships, half-sibships and large numbers of first cousins. However, such constellations of relatives may be infrequent in the population.
To test whether EPDs are less confounding of genetic and shared environmental effects than CTDs, and which design has greater statistical power to detect heritability, we used the Classic Mx statistical software package to compute simulations (Neale et al. 2004). We began with the proportion of 70 % A and 30 % E (this is a reasonable estimate for neuroimaging data of global brain measures; cf. Kremen et al. 2010). We then additionally used a proportion of 50 % A to examine power in the context of lower estimates. Power calculations were based on a simulated sample size of 486 twins (243 pairs) with an almost 50:50 split between MZ and DZ twins (122 MZ and 121 DZ pairs, respectively), and a simulated sample of pedigrees with 486 individuals. The total N of 486 was based on the N from an existing pedigree study, shortly to be described. This comparison is a conservative test of the CTD, because we treated all pairs of relatives in the pedigree data as if they were independent. Treating relative pairs as independent artificially increases the power of the EPD, because the many pairs of relatives are clearly not independent.
Large extended pedigree studies are rare. Here we refer to two such studies to provide some example of the proportion of different types of relatives: one with N = 486 (Glahn et al. 2010) and one with N = 397 (Souto et al. 2000). There were 18 and 30 % first-degree relatives, 18 and 34 % second-degree relatives, 27 and 26 % third-degree relatives, 24 and 9 % fourth-degree relatives, and 12 and 2 % fifth- or sixth-degree relatives.Footnote 1 Thus, first- and second-degree relatives comprised 36 % of one sample and 64 % of the other. In our simulated sample, we used 20 % first-degree relatives and 80 % second- through sixth-degree relatives.
Power calculations
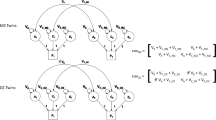
We represented the CTD and the EPD as structural equation models. These methods have been described in detail elsewhere (Martin and Eaves 1977; Satorra and Saris 1985; van der Sluis et al. 2008) and Mx code is provided in Supplement 1. Briefly, statistical power calculations for structural equation models may be obtained by: (i) simulating data based on a ‘true world’ model; (ii) fitting both the true world and the false model to these data; (iii) using twice the difference in log-likelihood between these two models (Δχ2) as a non-centrality parameter; and (iv) calculating the probability of rejecting the null hypothesis given a non-centrality parameter of this magnitude.
If we assume that C = 0, as is most often done in EPDs, the non-centrality parameter to test the hypothesis that A is also zero may be compared between the CTD and extended pedigree scenarios. For these twin data, the non-centrality parameter is 97.16 compared with 62.63 for the pedigree data. In other words, there is a larger Δχ2 value for the CTD than there is for the pedigree design when one fits the false model of A = 0. The larger χ2 value translates into greater statistical power. Here, we examined models constraining C and also allowing C to cause covariance among the pedigree relationships. Specifically, we used two variations of C parameterization: one in which C causes covariance among siblings only, and one in which C causes covariance between (i) siblings, (ii) parents and (iii) their children and spouses.
The left half of Table 1 shows that, when C is constrained to 0, the sample sizes required to achieve a given level of statistical power are always smaller for the CTD in comparison with the EPD. If C is free to cause covariance (i.e., it is included in the statistical model) the Δχ2 value for the twin data is 19.19, and it is 16.24 for the pedigree data. This suggests that the CTD is still more powerful when C is allowed to cause covariance. The right half of Table 1 shows the sample sizes required to achieve a given level of statistical power when C is allowed to cause covariance. When C is allowed to cause covariance, power to detect heritability is substantially reduced for both designs. The power advantage for the CTD over the EPD is reduced, but the Δχ2 value is still higher for the twin data. We also used a proportion of 50 % A to calculate power in the context of lower-than-typical heritability estimates for brain measures, with similar results (Table 2). These findings support the notion twin designs are generally more powerful than pedigree designs for detecting heritability.
In the primary model for the pedigree data, we assumed that C applied to both sibling and parent-offspring cases. If we assumed that C applied only to siblings in the EPD, its power to detect heritability would then be greater than that of the CTD. In that case, the Δχ2 value was 37.54 in comparison to the value of 19.19 for the twin data. Although these values may indicate that the EPD has greater power when C only affects sibling pairs, it must be noted that the EPD has been specified as 1966 independent pairs of relatives, whereas the pedigrees actually comprise only 486 individuals. Thus in this scenario, the power of the EPD is artificially inflated, and more accurate power estimation would require specification of the particular configurations of pedigrees.
Conclusion
The primary conclusion to be drawn from these power calculations is that under most scenarios, the CTD has greater power to estimate heritability than the EPD. Our comparison was a conservative test of the CTD, because we artificially increased the power of the pedigree design by treating pairs of relatives as if they were independent. The one situation in which power was greater in the EPD was under the assumption that shared environmental variance contributes to sibling similarity only.
Thus, our power calculations do lead to the conclusion that there would be less confounding of common environmental factors in the EPD, but this conclusion is only true given a key assumption that common environmental influences contribute only to sibling resemblances and not to other pairs of relatives. This is especially true in the examples provided where the EPD contained a relatively small proportion of siblings (10–15 %), while CPDs consist entirely of siblings. This assumption is of questionable validity for certain traits, such as those with parent–child environmental transmission [e.g., alcohol consumption (Kendler et al. 1994)], or household factors such as diet.
In practice, heritability estimates in non-twin extended pedigree studies have most often been based on the assumption that C = 0. The CTD has its most substantial power advantage over the EPD under this assumption. Furthermore, if one assumed that C = 0 at the outset, then the optimal MZ:DZ ratio in the CTD would be to recruit MZ twins only (Visscher 2004). If C = 0, then the environmental variance is simply estimated as 1 minus the MZ twin correlation (Dolan et al. 1999). Given this assumption, including MZ twins only (or at least a greater proportion of them) would further increase the power of the CTD to detect heritability.
Both designs have their strengths. The CTD can be advantageous with respect to age and cohort confounds. When twins are assessed at the same time, they are both the same age and from the same cohort. Thus both age and cohort effects do not affect their similarity. When either age x genotype or cohort x genotype interactions are present, simultaneous assessments of siblings will underestimate the effects of genotype or the shared environment, or both. For certain phenotypes, such as substance use, there is clear evidence of reduced resemblance between siblings the greater their difference in age (Verhulst et al. 2014).
On the other hand, an assumption of the CTD is that twins are representative of the general population. Although this appears to be the case for many phenotypes studied to date (Martin et al. 1997) it is not guaranteed for all phenotypes, and should be tested as a matter of routine. A further, frequently questioned, concern is the equal environment assumption (EEA) in CTDs. Although there is general empirical support for the EEA (Kendler et al. 1993; Loehlin and Nichols 1976), data to test it should be gathered in every new study and tested for every phenotype. To do this is relatively straightforward, by regressing intrapair phenotype differences on measures of similarity of environment.
In the present article, we found that the power of the extended pedigree design was greater when C is shared only by sib pairs as opposed to all members of nuclear families. In the examples provided, the EPD contains a relatively small proportion of siblings (10–15 %). By contrast, CPDs are entirely composed of siblings.
The analysis of sex-limitation can be less informative in a twin study than a study of, for example, siblings and half-siblings reared together. From a design perspective, MZ twins only occur as same-sex pairs, which means that only one form of the reduced across-sex correlation (rA or rC) can be parameterized. Since both siblings and half-siblings can occur in opposite-sex form, it is possible to fit a model that allows both variance components to be attenuated due to sex-limitation, through rA and rC.
Several additional analyses could further inform this research. First, the examination of gender distribution between designs is an important step. Also, testing more pedigree structures would further our understanding of the strengths and weaknesses of various pedigree designs in relation to twin studies.
Heritability remains an important construct. Maximally explaining heritability in a genome-wide association study (GWAS) is important, but heritability has also been referred to as the “dark matter” of the GWAS—we know it is there, but we have often been unable to “find” it (Manolio et al. 2009). Very large sample sizes are required to estimate heritability using restricted maximum likelihood estimation approaches in samples of unrelated individuals, and these estimates, to date, have been inconsistent with estimates from traditional twin and pedigree designs. Imprecise phenotypes are one important rate-limiting factor in GWAS designs. Overall, the degree of dissimilarity of estimates between genome-wide and family analyses has been unpredictable, and this issue is still unresolved. Studies of height, for example, provide more plausible heritability estimates using CTDs than using genome-wide complex trait analysis, or GCTA (Lango Allen et al. 2010; Yang et al. 2010; Visscher et al. 2010). We believe the CTD remains the initial method of choice for obtaining the most precise estimates of heritability as well as for determining optimal phenotype definitions. More precise heritability coefficients improve the accuracy of genetic correlations between traits, with obvious utility, e.g., in the analysis of MRI data (Chen et al. 2013).
Evidence suggests that twin-pedigree designs can offer the best of both worlds (Truett et al. 1994; Maes et al. 2009; Keller et al. 2010). Most significantly, it is important to avoid assuming that shared environmental factors play no role in a phenotype. We are not suggesting that models that exclude C (e.g., AE models) are not valid. Indeed, we frequently utilize such models ourselves. Rather, the point is that although certain traits might a priori be thought to be unaffected by environmental factors shared within families, it is optimal to proceed by first acquiring empirical evidence that they do not contribute to individual differences.
Notes
Numbers do not total to 100% due to rounding error.
References
Blangero J, Williams JT, Almasy L (2003) Novel family-based approaches to genetic risk in thrombosis. J Thromb Haemost 1:1391–1397
Carey G (2003) Human genetics for the social sciences. Sage Publications, Thousand Oaks
Chen CH, Fiecas M, Gutierrez ED, Panizzon MS, Eyler LT et al (2013) Genetic topography of brain morphology. Proc Natl Acad Sci USA 110:17089–17094
Coon H, Carey G (1989) Genetic and environmental determinants of musical ability in twins. Behav Genet 19:183–193
Dolan CV, Boomsma DI, Neale MC (1999) A note on the power provided by sibships of sizes 2, 3, and 4 in genetic covariance modeling of a codominant QTL. Behav Genet 29:163–170
Eaves LJ, Heath A, Martin N, Maes H, Neale M et al (1999) Comparing the biological and cultural inheritance of personality and social attitudes in the Virginia 30000 study of twins and their relatives. Twin Res. 2:62–80
Glahn DC, Winkler AM, Kochunov P, Almasy L, Duggirala R et al (2010) Genetic control over the resting brain. Proc Natl Acad Sci USA 107:1223–1228
Heath AC, Berg K, Eaves LJ, Solaas MH, Corey LA et al (1985) Educational policy and the heritability of educational attainment. Nature 314:734–736
Heath AC, Cates R, Martin NG, Meyer J, Hewitt JK et al (1993) Genetic contribution to risk of smoking initiation: comparisons across birth cohorts and across cultures. J Subst Abuse 5:221–246
Keller MC, Medland SE, Duncan LE, Hatemi PK, Neale MC et al (2009) Modeling extended twin family data I: description of the Cascade model. Twin Res. and Hum. Genet. 12:8–18
Keller MC, Medland SE, Duncan LE (2010) Are extended twin family designs worth the trouble? A comparison of the bias, precision, and accuracy of parameters estimated in four twin family models. Behav Genet 40:377–393
Kendler KS, Neale MC (2009) “Familiality” or heritability (letter to the editor). Arch Gen Psychiatry 66:452–453
Kendler KS, Neale MC, Heath AC, Kessler RC, Eaves LJ (1994) A twin-family study of alcoholism in women. Am J Psychiatry 151:707–715
Kendler KS, Neale MC, Kessler RC, Heath AC, Eaves LJ (1993) A test of the equal-environment assumption in twin studies of psychiatric illness. Behav Genet 23:21–27
Kendler KS, Neale MC, Sullivan P, Corey LA, Gardner CO et al (1999) A population-based twin study in women of smoking initiation and nicotine dependence. Psychol Med 29:299–308
Kremen WS, Prom-Wormley E, Panizzon MS, Eyler LT, Fischl B et al (2010) Genetic and environmental influences on the size of specific brain regions in midlife: the VETSA MRI study. Neuroimage 49:1213–1223
Lango Allen H, Estrada K, Lettre G, Berndt SI, Weedon MN et al (2010) Hundreds of variants clustered in genomic loci and biological pathways affect human height. Nature 467:832–838
Loehlin JC, Nichols RC (1976) Heredity, environment and personality: a study of 850 sets of twins. University of Texas Press, Austin
Maes HH, Neale MC, Medland SE, Keller MC, Martin NG et al (2009) Flexible Mx specification of various extended twin kinship designs. Twin Res Human Genet 12:26–34
Manolio TA, Collins FS, Cox NJ, Goldstein DB, Hindorff LA et al (2009) Finding the missing heritability of complex diseases. Nature 461:747–753
Martin NG, Eaves LJ (1977) The genetical analysis of covariance structure. Heredity 38:79–95
Martin N, Boomsma D, Machin G (1997) A twin-pronged attack on complex traits. Nat Genet. 17:387–392
Neale MC, Cardon LR (1992) Methodology for genetic studies of twins and families. Kluwer Academic Publishers, Dordrecht
Neale MC, Boker SM, Xie G, Maes HH (2004) Mx: Statistical Modeling, 6th edn. Department of Psychiatry, Medical College of Virginia, Richmond
Posthuma D, Boomsma DI (2000) A note on the statistical power in extended twin designs. Behav Genet 30:147–158
Satorra A, Saris WE (1985) Power of the likelihood ratio test in covariance structure analysis. Psychometrika 54:131–151
Schork NJ, Schork MA (1993) The relative efficiency and power of small-pedigree studies of the heritability of a quantitative trait. Hum Hered 43:1–11
Souto JC, Almasy L, Borrell M, Gari M, Martinez E et al (2000) Genetic determinants of hemostasis phenotypes in Spanish families. Circulation 101:1546–1551
Truett KR, Eaves LJ, Walters EE, Heath AC, Hewitt JK et al (1994) A model system for analysis of family resemblance in extended kinships of twins. Behav Genet 24:35–49
van der Sluis S, Dolan CV, Neale MC, Posthuma D (2008) Power calculations using exact data simulation: a useful tool for genetic study designs. Behav Genet 38:202–211
Verhulst B, Eaves LJ, Neale MC (2014) Moderating the covariance between family members’ substance use behavior. Behav Genet 44:337–346
Visscher PM (2004) Power of the classical twin design revisited. Twin Res. 7:505–512
Visscher P, Hill WG, Wray NR et al (2008) Heritability in the genomics era—concepts and misconceptions. Nat Rev Genet 9:255–266
Visscher PM, Yang J, Goddard MEA et al (2010) A commentary on ‘common SNPs explain a large proportion of the heritability for human height’ by Yang et al (2010). Twin Res Human Genet 13:517–524
Williams JT, Blangero J (1999) Power of variance component linkage analysis to detect quantitative trait loci. Ann Hum Genet 63:545–563
Winkler AM, Kochunov P, Blangero J, Almasy L, Zilles K et al (2009) Cortical thickness or grey matter volume? The importance of selecting the phenotype for imaging genetics studies. Neuroimage 53(3):1135–1146
Yang J, Benyamin B, McEvoy BP, Gordon S, Henders AK et al (2010) Common SNPs explain a large proportion of the heritability for human height. Nat Genet 42:565–569
Acknowledgments
This work was supported by National Institute on Aging (AG022381, AG018384, AG018386, AG022982); National Center for Research Resources (P41-RR14075; NCRR BIRN Morphometric Project BIRN002); National Institute for Biomedical Imaging and Bioengineering (R01EB006758); National Institute for Neurological Disorders and Stroke (R01 NS052585-01); Mental Illness and Neuroscience Discovery (MIND) Institute, part of the National Alliance for Medical Image Computing (NAMIC), funded by the National Institutes of Health through the NIH Roadmap for Medical Research, Grant U54 EB005149. Additional support was provided by The Autism and Dyslexia Project funded by the Ellison Medical Foundation. The U.S. Department of Veterans Affairs has provided financial support for the development and maintenance of the Vietnam Era Twin (VET) Registry. Numerous organizations have provided invaluable assistance in the conduct of this study, including: Department of Defense; National Personnel Records Center, National Archives and Records Administration; Internal Revenue Service; National Opinion Research Center; National Research Council, National Academy of Sciences; the Institute for Survey Research, Temple University. Most importantly, the authors gratefully acknowledge the continued cooperation and participation of the members of the VET Registry and their families. Without their contribution this research would not have been possible.
Conflict of interest
Anna R. Docherty, William S. Kremen, Matthew S. Panizzon, Elizabeth C. Prom-Wormley, Carol E. Franz, Michael J. Lyons, Lindon J. Eaves and Michael C. Neale declare that they have no conflicts of interest to declare.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human participants or animals performed by any of the authors. Informed consent does not apply.
Author information
Authors and Affiliations
Corresponding author
Additional information
Edited by Matt McGue.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Docherty, A.R., Kremen, W.S., Panizzon, M.S. et al. Comparison of Twin and Extended Pedigree Designs for Obtaining Heritability Estimates. Behav Genet 45, 461–466 (2015). https://doi.org/10.1007/s10519-015-9720-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10519-015-9720-z