
Abstract
Proline/arginine-rich end and leucine-rich protein (PRELP) is identified as a small proteoglycan in the extracellular matrix that has been tightly associated with cell adhesion. At present, the role of PRELP in colorectal cancer (CRC) remains largely unknown. PRELP expression in human CRC tissue samples was analyzed by qRT-PCR and immunochemistry. CCK-8, colony formation, transwell, and tube formation assays were utilized to determine the influences of PRELP on the malignant phenotypes of CRC cells. Mouse xenograft and tumor metastasis models were constructed to further validate the function of PRELP. Furthermore, we investigated the efficacy of PRELP combined with bevacizumab treatment in a mouse xenograft model of CRC. Additionally, RNA-seq was performed to analyze the potential signaling pathways regulated by PRELP. Immunofluorescence staining and coimmunoprecipitation were conducted to confirm the interaction between PRELP and fibroblast growth factor 1 (FGF1). In this study, we found that PRELP exerted a tumor-suppressive effect on CRC. The expression level of PRELP was significantly reduced in CRC tissues and cell lines. Both in vivo and in vitro experiments confirmed that PRELP inhibited CRC cell proliferation, promoted apoptosis, and suppressed migration and invasion via a reduction in the epithelial-mesenchymal transition and attenuated angiogenesis, thereby dampening tumor progression. In addition, PRELP markedly potentiated the efficacy of bevacizumab in a mouse xenograft model. Mechanistically, PRELP bound to FGF1 and reduced the stability of the FGF1 protein, accompanied by an increase in its degradation, which subsequently inactivated the PI3K/AKT/mTOR pathway, thereby leading to reduction in tumor angiogenesis and metastasis. Our study for the first time unveiled the tumor-suppressive role of PRELP in CRC and provided a potential effective strategy for the treatment of CRC.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Colorectal cancer (CRC) is one of the most common malignant tumors worldwide and ranks third in terms of morbidity and mortality rates [1]. CRC patients often have a poor prognosis due to tumor recurrence and metastasis [2,3,4,5]. Epithelial-mesenchymal transition (EMT) and angiogenesis have been considered as the main causes of CRC progression [6, 7]. Although antiangiogenic therapy is currently the standard treatment for CRC, its efficacy is still limited [8]. Therefore, new strategies are urgently needed to improve the antitumor efficacy.
Proline/arginine-rich end and leucine-rich protein (PRELP) is a member of the family of small leucine-rich proteoglycans (SLRPs) characterized by N-terminal structural domains with proline- and arginine-rich residues [9]. PRELP binds to collagen in connective tissues through its LRR structural domain [10], and its N-terminus also binds to heparin and acetyl heparin sulfate proteoglycans in the extracellular matrix (ECM) [11], thereby contributing to the maintenance of the cellular structure and tissue homeostasis [12]. PRELP has been shown to be suppressed in many cancers [13,14,15,16,17], such as hepatocellular carcinoma and bladder cancer, which exerts a tumor-suppressive function via a reduction in the EMT. A recent study indicated that PRELP suppresses melanoma progression by enhancing the immunoreactivity of CD8+ T cells [18]. However, to date there is a lack of research on the role of PRELP in CRC, we thus explored the function of PRELP in CRC in this study.
Fibroblast growth factor 1 (FGF1) is one of the secreted proteins in the fibroblast growth factor (FGF) family. FGF1 regulates important processes such as cell proliferation, migration, and differentiation by activating specific tyrosine kinase receptors on the cytomembrane [19]. Upon binding to the FGF receptor (FGFR), FGF1 activates intracellular tyrosine kinase structural domains to initiate cascade signaling, including the RAS/MAPK and PI3K/AKT pathways [20]. Studies have shown that FGF1 promotes tumor growth and angiogenesis [21, 22], thereby facilitating tumor progression. Currently, there is no information on the association of PRELP with FGF1.
In this study, we found that PRELP inhibited CRC cell proliferation, migration, invasion, EMT, and angiogenesis. Specifically, PRELP could bind with FGF1 to decrease the stability of FGF1, which subsequently suppressed the PI3K/AKT/mTOR signaling pathway, thereby leading to reductions in CRC angiogenesis and metastasis. Our findings provide new insights into the inhibitory role of PRELP in CRC progression and help to reassess its potential value for CRC treatment.
Materials and methods
Human tissue samples, cell lines, and drugs
This study was approved by the Ethics Committee of the First Affiliated Hospital of Chongqing Medical University (No. 2023–222) and complied with the Declaration of Helsinki. Informed consent was obtained from all patients included in this study. All the samples were clinically and pathologically diagnosed. The CRC tumor tissues and paired normal tissues used in this study were obtained from the First Affiliated Hospital of Chongqing Medical University.
Human colorectal cancer cell lines (SW480, SW620, HCT116, Lovo, and RKO), human normal colorectal epithelial cells (NCM460), and human umbilical vein endothelial cells (HUVECs) were purchased from the American Type Culture Collection. All cells were cultured in DMEM (Gibco, USA) supplemented with 10% fetal bovine serum (Gibco, USA) and 1% penicillin–streptomycin (Gibco, USA). All cells were cultured in an incubator at 37 °C with 5% CO2 and free from mycoplasma contamination.
The PI3K activator 740 Y-P (20 µM, HY-P9906A, MedChemExpress, USA), inhibitor LY294002 (10 µM, HY-10108, MedChemExpress, USA), MG132 (10 µM, HY-13259, MedChemExpress, USA), and Cycloheximide (CHX, 50 µg/ml, HY-12320, MedChemExpress, USA) were dissolved in DMSO for cell culture. The FGF1 protein (10 ng/ml, HY-P7001, MedChemExpress, USA) was dissolved in PBS.
RNA extraction and quantitative real-time PCR (qRT-PCR)
The cells were lysed in TRIzol (Takara, Japan), and RNA was extracted. According to the manufacturer’s instructions, cDNA was synthesized using a reverse transcription kit (Accurate Biology, China). The SYBR Green Premix Pro Taq HS qPCR Kit (Accurate Biology, China) was used for qRT-PCR. We evaluated the relative expression levels of genes using the 2−△△CT method. GAPDH was used as an internal control. The sequences of primers used in this study were listed in the Supplementary file.
Cell transfection and construction of stable cell lines
PRELP was inserted into the pcDNA3.1( +) vector to construct an overexpression plasmid, and the cells were transfected with the aid of Lipofectamine 2000 (Invitrogen, USA) according to the manufacturer’s instructions. The transfected cells were screened with G418 to construct a cell line stably expressing PRELP. The shRNAs targeting PRELP (sh-PRELP-1, 5’-CCGGGGATTAACCTGGACAACAACCCTCGAGGGTTGTTGTCCAGGTTAATCCTTTTTT-3’; sh-PRELP-2, 5’-CCGGGGAGAAGAACCAGTTGGAAGACTCGAGTCTTCCAACTGGTTCTTCTCCTTTTTT-3’; sh-PRELP-3, 5’-CCGGGCCCACAACATCCTGAGAAAGCTCGAGCTTTCTCAGGATGTTGTGGGCTTTTTT-3’; and sh-PRELP-4, 5’-CCGGGCTTTCCCAATCTTGCCTTCACTCGAGTGAAGGCAAGATTGGGAAAGCTTTTTT-3’) were acquired from Tsingke (Tsingke Biotechnology, China).
Enzyme-linked immunosorbent assay (ELISA)
ELISA was performed according to the methodology published in previous studies [23]. SW620 and RKO cells stably expressing PRELP and controls were incubated in Petri dishes with complete medium supplemented with 2% FBS for 48 h. Supernatants were collected and centrifuged, and the concentration of secreted PRELP was measured using the Human PRELP ELISA Kit (EH3648, FineTest, China) according to the manufacturer's instructions.
Immunohistochemistry (IHC) and Immunofluorescence (IF) analyses
Formalin-fixed and paraffin-embedded tissue sections were deparaffinized, peroxidase activity was quenched, and the sections were blocked and incubated overnight at 4 °C with a PRELP antibody (#40,410, 1:100, Signalway Antibody, USA). The sections were incubated with a secondary antibody at 37 °C for 30 min. The target proteins were visualized via DAB staining of the bound antibodies (K176810E, ZSGB-BIO, China), and the nuclei were counterstained with hematoxylin. Images of immunostained sections were acquired with a Leica microscope (Leica Microsystems, Germany). Scoring of the staining intensity was performed independently by two investigators. Tissue staining was evaluated using the immune response score (IRS). The staining intensity was scored from 0 to 3: 0, no staining; 1, low staining; 2, moderate staining; and 3, high staining. The percentage of positive cells ranged from 0 to 4 points: 0, < 5%; 1, 6%—25%; 2, 26%–50%; 3, 51%–75%; and 4, > 80%. The final score was obtained by multiplying the staining intensity and the percentage of positive cells.
Immunofluorescence staining was performed as previously described [24]. The tissue sections were incubated overnight at 4 °C with the following primary antibodies: PRELP (23,783–1-AP, 1:100, Proteintech, China) and FGF1 (sc-55520, 1:50, Santa Cruz, USA). DAPI (Beyotime, China) was used to restain the tissue before sealing. Photographs were captured with a confocal laser scanning microscope.
Cell Counting Kit-8 (CCK-8) and colony formation assays
For the CCK-8 experiments, cells were seeded at a density of 2 × 103 cells per well in 96-well plates. CCK-8 reagent (C0005, TargetMol, USA) was added at 0 h, 24 h, 48 h, and 72 h to assess cell viability. For the colony formation assays, 1 × 103 cells were added to each well of a 6-well plate and cultured for approximately 2 weeks to observe colony formation. The cells were fixed with 4% paraformaldehyde (Biosharp, China) and stained with a 0.1% crystal violet solution (Beyotime, China).
Transwell assays
Cell migration and invasion abilities were assayed in 24-well plates using 8 µm pore-size chambers (Corning, USA) coated with or without Matrigel (BD Biosciences, USA). Transwell assays were performed as previously described [25]. Fixation and staining were performed after 48 h, and the cells that migrated to the lower side of the chambers were photographed using a Leica microscope.
Flow cytometry
The flow cytometry assay was performed according to the methods published in previous studies [26]. The cells to be tested were digested, collected, and washed. The cells were stained with Annexin V-FITC and propidium iodide to detect apoptosis. For cell cycle detection, the cells were fixed with 75% ethanol and then stained sequentially with propidium iodide. The data were collected using a flow cytometer (BD Biosciences, USA).
Western blot and coimmunoprecipitation (Co-IP)
Total cellular protein was extracted with cell lysis buffer containing protease and phosphatase inhibitors (Beyotime, China). Proteins were quantified using the BCA protein quantification kit (Beyotime, China). Electrophoresis (SDS‒PAGE) was performed on a 10% polyacrylamide gel, after which the proteins were transferred to a PVDF membrane. The membranes were blocked for 2 h with 5% skim milk and then incubated overnight at 4 °C with primary antibodies. Primary antibodies against the following targets were used: PRELP (23,783–1-AP, 1:1000, Proteintech), BCL-2 (#2870, 1:1000, Cell Signaling Technology), BCL-XL (#2764, 1:1000, Cell Signaling Technology), BAD (#9292, 1:1000, Cell Signaling Technology), BAX (WL01637, 1:1000, Wanleibio), cleaved caspase 3 (#9661, 1:1000, Cell Signaling Technology), E-cadherin (20,874–1-AP, 1:20,000, Proteintech), N-cadherin (66,219–1-Ig, 1:5000, Proteintech), Vimentin (10,366–1-AP, 1:2000, Proteintech), Snail (13,099–1-AP, 1:1000, Proteintech), MMP2 (#40,994, 1:1000, Cell Signaling Technology), MMP9 (#13,667, 1:1000, Cell Signaling Technology), Claudin1 (WL03073, 1:1000, Wanleibio), PI3K (WL02240, 1:1000, Wanleibio), AKT (WL0003b, 1:1000, Wanleibio), PTEN (22,034–1-AP, 1:2000, Proteintech), p-PI3K (YP0224, 1:1000, Immunoway), p-AKT (#4060, 1:1000, Cell Signaling Technology), mTOR (ab134903, 1:10,000, Abcam), p-mTOR (ab109268, 1:5000, Abcam), and GAPDH (60,004–1-Ig, 1:50,000, Proteintech). The next day, the membranes were incubated for 1 h with HRP-conjugated secondary antibodies. Final visualization was performed with a Western blot kit (WBKLS0100, MerckMillipore, USA) and an enhanced chemiluminescence system.
For the Co-IP assay, cellular protein extracts containing PRELP fused with Flag tags were incubated overnight at 4 °C with anti-Flag magnetic beads (HY-K0207, MedChemExpress, USA) and anti-IgG (AC011, ABclonal, China). Then, the magnetic beads were purified, and the Co-IP complexes were examined by Western blot.
RNA sequencing (RNA‑seq)
Total RNA was extracted by lysing cells stably expressing PRELP and blank load-transfected cells using TRIzol (Takara, Japan), and processing and RNA sequencing were subsequently performed by BGI Tech (Shenzhen, China). Single-ended 50 bp reads were generated on the BGIseq500 platform (BGI-Shenzhen, China). DEGs were visualized in volcano plots using the ggplot2 package. Heatmaps were generated with the pheatmap package. GO and KEGG analyses were performed using the DAVID database (https://david.ncifcrf.gov/). GSEA of the DEGs was performed with GSEA 4.3.2 software.
Tube formation assay
Matrigel (BD Biosciences, USA) was diluted and inoculated in 96-well plates at 50 µl per well. When the Matrigel solidified, the appropriate density of HUVECs was inoculated into the corresponding wells, and different cell culture supernatants were added to a volume of 200 µl. The cells were incubated in an incubator with 5% CO2 at 37 °C for approximately 4 h. The angiogenic capacity was assessed by counting the number of tubes and branches under a microscope.
Sphere formation assay
SW620 and RKO cells were inoculated in ultralow attachment surface 6-well plates (Corning, USA) at 2 × 103 cells/well. The cells were incubated in DMEM/F12 (Gibco, USA) containing 20 ng/ml EGF (AF-100–15, Peprotech, USA), 10 ng/ml bFGF (100-8b, Peprotech, USA), and 10 µl/ml B27 (17,504,044, Gibco, USA) for approximately 2 weeks. Sphere formation was observed under an inverted microscope and photographs were captured (Leica Microsystems, Germany).
In vivo experiments
The Experimental Animal Management and Use Committee of Chongqing Medical University approved the experiment (approval number: IACUC-CQMU-2024–0269). Methods were performed according to the approved guidelines. The BALB/c nude mice (5 weeks old, female, 14–16 g) used in this study were obtained from Beijing Vital River Experimental Animal Technology (Beijing, China). For the subcutaneous xenograft tumor model, 1 × 106 cells were suspended in 100 μl of PBS and injected subcutaneously into both sides of the mice (n = 6). Tumor growth was monitored every two days starting at 7 days after injection. For the bevacizumab-treated model, when the volume of the subcutaneous tumors reached approximately 100 mm3, the nude mice were randomly divided into four groups (vector, PRELP, vector + bevacizumab, and PRELP + bevacizumab; n = 5). Intraperitoneal injections of 5 mg/kg bevacizumab (HY-P9906A, MedChemExpress, USA) or PBS were administered twice weekly. All the mice were sacrificed after being monitored for 21 days. The obtained tumor tissues were weighed and embedded in paraffin. The tumor volume was calculated as follows: tumor volume = (length × width2)/2.
For the metastatic tumor model, cells stably overexpressing PRELP or vector (1 × 106 cells in 100 μl of PBS per mouse) were injected into the tail vein or spleen, respectively, of nude mice randomly assigned to groups (n = 3). For mice requiring transsplenic injection, after anesthesia was administered by an intraperitoneal injection of 1% pentobarbital sodium (50 mg/kg), the abdominal skin of the mice was sterilized, and an incision of approximately 1 cm was made. After the spleen was exposed, 100 μl of cells were slowly injected into the spleen. The spleen was gently placed back into the abdominal cavity, and then the abdominal wall and skin were sutured. The activity and mental condition of the nude mice were observed. When the condition of the mice deteriorated (45 days after injection), the mice were sacrificed. Intact liver and lung tissues were isolated from the mice. The number of metastatic nodules was counted and photographed.
Statistical analysis
All the statistical analyses were performed using GraphPad Prism 9.0 and SPSS 25.0 software. Categorical data are presented as numbers, which were compared using the chi-square test or Fisher’s exact test. Student’s t-test or one-way ANOVA was used to assess the significance of differences in continuous variables between groups. The data were obtained from at least three independent experiments. P-values < 0.05 were considered statistically significant. * P < 0.05, ** P < 0.01, *** P < 0.001, and **** P < 0.0001.
Results
PRELP expression was significantly reduced in CRC tissues and cell lines and related to the prognosis of CRC patients
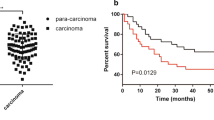
Considering the important role of ECM in tumors, we performed differential analysis of the EXTRACELLULAR_REGION gene set from the GSEA database in the RNA-seq data for CRC and normal tissues from TCGA database. Among the genes with the most significant differences (|Log2FC|> 2, P < 0.05), we chosed the top-ranked and less-studied PRELP to further explore (Fig. 1A). To clarify the expression of PRELP in tumors, we searched the TIMER 2.0 database for PRELP expression across cancers, and the results suggested that PRELP was expressed at a low level in most cancers (Fig. 1B). We compared the expression of PRELP using RNA-seq data for paired CRC and normal samples (n = 41) from TCGA database, and the results indicated that PRELP expression was reduced in CRC (Fig. 1C). Next, we analyzed the PRELP protein expression levels in CRC (n = 97) and normal tissues (n = 100) in the UALCAN database (Fig. 1D). We analyzed 18 paired fresh CRC tissue samples via qRT-PCR to verify these findings. The results revealed that PRELP was frequently downregulated in CRC tissues compared with normal tissues (Fig. 1E). Moreover, we further collected 40 paired formalin-fixed and paraffin-embedded CRC tissue sections to detect PRELP expression viaIHC. IHC showed similar results (Fig. 1F). Notably, we observed that PRELP was more abundant in the ECM of the colonic glandular ducts (Fig. 1F). Those 40 CRC patients were divided into two groups according to the IRS: high PRELP expression (n = 23) and low PRELP expression (n = 17). Compared with patients with high PRELP expression, patients with low PRELP expression had more advanced tumor stages and metastasis (Table 1). We followed up the survival data of these CRC patients. The overall survival analysis showed that the high PRELP expression group improved a significantly better prognosis than low PRELP expression group (Fig. 1G). We also examined PRELP expression in normal colorectal epithelial cells NCM460 and common colorectal cancer cell lines using qRT-PCR and Western blot. The results revealed that its expression was significantly reduced in CRC cell lines, especially in SW620 and Lovo cells, which have high metastatic potential, and in the poorly differentiated RKO cells (Fig. 1H–I). Consequently, PRELP expression was decreased in CRC tissues and cell lines and related to the prognosis.

PRELP is expressed at low levels in CRC and related to the prognosis of CRC patients. A Volcano plot of genes in the EXTRACELLULAR_REGION gene set which are differentially expressed in CRC and normal tissues from TCGA database. B The expression levels of PRELP in pan-cancer were analyzed using the TIMER 2.0 database. C PRELP mRNA expression in paired CRC and normal tissue samples in TCGA database. D Expression levels of PRELP in CRC and normal tissues in the ULCAN database. E qRT-PCR analysis of PRELP mRNA expression in 18 pairs of primary CRC tissues and adjacent normal tissues. F Representative IHC images of 40 pairs of CRC and normal tissue samples and statistical plots of the IRS score. G Overall survival analysis of 40 patients. H qRT-PCR analysis of the PRELP mRNA expression levels in NCM460 and CRC cell lines. I Western blot analysis of PRELP protein levels in NCM460 and CRC cell lines. * P < 0.05, ** P < 0.01, and *** P < 0.001
PRELP inhibited CRC cell proliferation, promoted apoptosis in vitro and suppressed tumorigenesis in the xenograft model
To investigate the effect of PRELP on the malignant phenotype of CRC cells, we selected SW620 and RKO cells with low PRELP expression to construct cell lines stably overexpressing PRELP, as determined by Western blot (Fig. 2A). Since PRELP is a secreted protein, we further collected SW620 and RKO cell supernatants for ELISA to confirm the increased level of PRELP in the extracellular space. (Fig. 2B). To investigate the effect of PRELP on CRC cell proliferation, we performed CCK-8 proliferation assays and colony formation assays and the results revealed that PRELP inhibited the proliferation of CRC cells (Fig. 2C and 2D). An analysis of the cell cycle distribution by flow cytometry revealed that PRELP halted SW620 in the G2 phase and halted RKO in G1 phase (Fig. 2E). Furthermore, we analyzed the cell apoptosis rate by flow cytometry and found that PRELP promoted the apoptosis of CRC cells (Fig. 2F). These results were further corroborated through the decreased expression of Bcl-2 and Bcl-XL and the increase in Bad, Bax, and cleaved Caspase-3 expression by Western blot (Fig. 2G). To investigate the effect of PRELP on cell proliferation in vivo, we subcutaneously injected nude mice with SW620 cells stably overexpressing PRELP. Compared with the vector group, the PRELP-overexpressing group exhibited slower tumor growth and a smaller volume (Fig. 2H and I). IHC was performed on xenograft tissue sections, which displayed lower expression of Ki-67 in the PRELP group than in the vector group (Fig. 2J). Altogether, these findings indicated that PRELP inhibited the proliferation and promoted the apoptosis of CRC cells in vivo and in vitro.

PPRELP inhibited CRC cell proliferation in vivo and in vitro. A-B Stable overexpression of PRELP in SW620 and RKO cells was detected using Western blot (A) and ElISA (B). C, D Cell proliferation was analyzed using the CCK-8 assay (C) and colony formation assay (D). E Cell cycle alterations were measured using flow cytometry in cells stably expressing the PRELP and the control vectors. F The proportions of apoptotic cells in the PRELP-overexpressing and vector groups were assessed. Histograms shows changes in the percentages of apoptotic cells. G Western blot analysis of the levels of apoptosis-related molecules in SW620 and RKO cells stably expressing PRELP or the vector. Histograms showed changes in the gray values of the Western blot. H-J SW620 cells stably overexpressing PRELP or the vector were injected subcutaneously into nude mice. Xenografts were collected after 21 days (H), tumor growth curves were plotted, and tumor weights were measured (I). H&E and IHC staining were performed (J) * P < 0.05, ** P < 0.01, and *** P < 0.001
PRELP suppressed CRC cell migration, invasion, stemness, and angiogenesis in vitro and reduced liver and lung metastasis by inhibiting the EMT
We stained the cytoskeleton with phalloidin. Cytoskeletal rearrangement and microfilament protein disorganization were significantly observed in the PRELP group, in which the cell morphology changed from a shuttle shape to an obtuse round shape, (Fig. 3A). We further detected the effects of PRELP on CRC cell migration and invasion. The results of the transwell assay revealed that PRELP could reduce the number of SW620 and RKO cells (Fig. 3B). Moreover, we detected the protein expression of EMT-related markers via Western blot. We observed increased E-cadherin expression and decreased expression of N-cadherin, Vimentin, MMP 2, MMP 9, Snail, and Claudin 1, suggesting a reduction in the EMT (Fig. 3C). Since angiogenesis is an important factor in tumor metastasis, we performed a tube formation assay to assess the effect of PRELP on angiogenesis. The results revealed that PRELP markedly inhibited tube formation by HUVECs (Fig. 3D). The metastatic ability of tumors is also associated with the stemness of tumor cells. Therefore, we analyzed the expression of CD44, CD24, CD133, and NANOG via qRT-PCR, which revealed that PRELP inhibited the expression of the above stemness-related genes (Fig. 3E). Cell sphere formation assays confirmed that PRELP decreased the number of spheres formed by SW620 and RKO cells (Fig. 3F). To verify PRELP attenuated CRC metastasis, we constructed a mouse metastatic tumor model. Two groups of BALB/c nude mice received intrasplenic injections of SW620 cellsstably overexpressingPRELP or the vector, which were used to assess liver metastasis. Moreover, two additional groups of mice were evaluated for the lung metastatic potential by a tail vein injection. The tumor metastasis model and H&E staining revealed that the number and volume of tumor nodules in the liver and lungs of the PRELP group were fewer and smaller than those in the control group(Fig. 3G–H). To explore the role of PRELP from the other side, we selected SW480 and HCT116 cells with relatively high PRELP expression levels to construct cell lines with stable knockdown of PRELP (Fig. 4A). CCK-8 and colony formation assays showed that the knockdown of PRELP promoted the proliferation of CRC cells (Fig. 4B–C). PRELP knockdown alsoobviously promoted the migration and invasion of SW480 and HCT116 cells, as shown by transwell assays (Fig. 4D). Similarly, we found that sh-PRELP increased the expression of EMT-promoting markers (Fig. 4E). Taken together, these results indicated that PRELP could inhibit CRC metastasis by suppressing the EMT.

PRELP inhibited CRC cell migration, invasion, stemness, EMT, and metastasis. A Cells stably overexpressing PRELP and control cells were stained using phalloidin to visualize alterations in the cytoskeletal morphology. B CRC cells stably expressing the vector or PRELP were subjected to transwell assays. C Western blot analysis of EMT-related markers in SW620 and RKO cells overexpressing PRELP or the vector. D Cell culture supernatants from the vector and PRELP groups were added to HUVECs to detect tube formation ability. E qRT-PCR analysis of stemness markers in cells stably expressing the vector or PRELP. F Sphere formation assay of SW620 and RKO cells overexpressing PRELP or the vector. G-H SW620 cells stably overexpressing PRELP or the vector were injected into the tail vein or spleen of nude mice, and representative images of lung metastasis, liver metastasis, and nodule statistics (G) as well as representative images of H&E-stained lungs and livers (H), are shown. * P < 0.05, ** P < 0.01, and *** P < 0.001

Knockdown of PRELP enhances CRC cell proliferation, migration, and invasion. A Western blot analysis was performed to detect the stable knockdown of PRELP expression in SW480 and HCT116 cells. B-C Cell proliferation was assessed using a CCK-8 proliferation assay (B) and a colony formation assay (C). D Migration and invasion were evaluated in cells transfected with sh-NC or sh-PRELP via transwell assays. E Western blot analysis of EMT-associated markers in SW480 and HCT116 cells with stable knockdown of PRELP or the control. * P < 0.05, ** P < 0.01, and *** P < 0.001
PRELP inactivated the PI3K/AKT/mTOR signaling pathway
To explore the mechanism by which PRELP regulates the malignant phenotype of CRC, transcriptome sequencing was performed on PRELP-overexpressing and control SW620 and RKO. A total of 2675 DEGs, including 1105 upregulated DEGs and 1570 downregulated DEGs, were identified in SW620 cells. A total of 1465 DEGs, including 1085 upregulated DEGs and 380 downregulated DEGs, were identified in RKO cells (Fig. 5A–B). We performed an enrichment analysis of these DEGs. The GO enrichment results for both cell lines revealed that the DEGs were related to signal transduction, cell adhesion, angiogenesis, and the regulation of apoptosis (Fig. 5C). Interestingly, the KEGG enrichment analysis revealed that the DEGs in the two cell lines were highly enriched in the PI3K/AKT signaling pathway (Fig. 5D). GSEA indicated that PRELP expression was negatively correlated with the PI3K/AKT signaling pathway (Normalized ES: -1.4314) (Fig. 5E). Therefore, we continued to explore changes in the PI3K/AKT pathway. Western blot was used to analyze the major markers of the PI3K/AKT pathway, which suggested that PRELP expression could decrease PI3K, AKT, and mTOR phosphorylation and increase PTEN expression, indicating the inhibition of the signaling pathway (Fig. 5F). VEGFA is a protein that promotes angiogenesis and is a downstream gene regulated by the PI3K/AKT pathway. We found that PRELP could decrease VEGFA levels via western blot. This finding was consistent with our experimental results for the tube formation assays described above. In addition, we demonstrated that PRELP knockdown promoted the activation of the PI3K/AKT pathway in HCT116 and SW480 cells, which further validated these results (Fig. 5G). Accordingly, the results suggested that PRELP inhibited CRC angiogenesis and tumor progression by suppressing the PI3K/AKT/mTOR signaling pathway.

PRELP inhibits the PI3K/AKT signaling pathway and leads to the suppression of the EMT and angiogenesis. A-B SW620 and RKO cells stably expressing the vector or PRELP were subjected to RNA-seq, and the volcano plots (A) and heatmaps (B) show the DEGs obtained. C-E GO enrichment analysis (C) KEGG enrichment analysis (D) and GSEA (E) were performed for DEGs obtained from SW620 and RKO cells. F The expression of PI3K/AKT/mTOR pathway proteins in SW620 and RKO cells stably expressing the vector or PRELP was detected via Western blot. Histograms showing the changes in gray values. G PI3K/AKT pathway protein expression in SW480 and HCT116 cells transfected with sh-NC or sh-PRELP was detected using Western blot. Histograms showing altered gray values of the immunoblots. * P < 0.05, ** P < 0.01, and *** P < 0.001
PRELP attenuated tumor angiogenesis and metastasis by downregulating the PI3K/AKT/mTOR signaling pathway
To verify that PRELP acted mainly through the PI3K/AKT/mTOR signaling pathway, we added the PI3K activator 740 Y-P to the cell culture medium. CCK-8 proliferation assays revealed that the addition of 740 Y-P partially restored the proliferative capacity of SW620 and RKO cells, which was previously inhibited by the overexpression of PRELP (Fig. 6A). Transwell assays revealed that CRC cells regained the ability of migration and invasion by 740 Y-P (Fig. 6B). Tube formation assays confirmed that tube formation ability, which was inhibited by PRELP, was increased by the addition of a PI3K activator (Fig. 6C). Furthermore, the addition of 740 Y-P promoted the phosphorylation of PI3K, AKT, and mTOR, which was inhibited by PRELP, and attenuated the expression of PTEN, indicating that pathways were activated. We analyzed EMT-related markers using Western blot, and the results showed that the PI3K activator could partially increase the expression of N-cadherin, vimentin, MMP2, and MMP9, while the level of E-cadherin was reduced. We also found that the addition of 740 Y-P rescued VEGFA expression, promoting angiogenesis (Fig. 6D). We further verified the above conclusions by adding the pathway inhibitor LY294002 to SW480 and HCT116 cells in the sh-PRELP and sh-NC groups. The inclusion of the inhibitor significantly suppressed the promotion of CRC cell growth, migration, invasion, and angiogenesis caused by PRELP knockdown (Fig. 6E–H). These findings suggested PRELP regulated angiogenesis and metastasis through the PI3K/AKT/mTOR signaling pathway.

PRELP inhibited the PI3K/AKT/mTOR signaling pathway and reduced the proliferation, metastasis, and angiogenesis of CRC cells. A-C Effects on the proliferation, migration, invasion, and angiogenesis of SW620 and RKO cells were detected via a CCK-8 assay (A) Transwell assay (B) and tube formation assay (C) after the addition of the PI3K activator 740 Y-P to the cell culture medium. D Changes in the expression of PI3K/AKT/mTOR pathway molecules, EMT markers, and VEGFA after the addition of 740 Y-P were detected using Western blot. Histograms show the altered gray values of the immunoblots. E–G CCK-8 (E) Transwell (F) and tubule formation (G) assays were used to analyze the effects of the PI3K inhibitor LY294002 with sh-NC or sh-PRELP on proliferation, migration, invasion, and angiogenesis of SW480 and HCT116. H Changes in the expression of PI3K/AKT/mTOR pathway proteins, EMT markers, and VEGFA in response to LY294002, as determined by Western blot. * P < 0.05, ** P < 0.01, and *** P < 0.001
PRELP inactivated the PI3K/AKT/mTOR signaling pathway by binding to FGF1 and reducing the stability of the FGF1 protein
FGF1, a potent regulator of the PI3K/AKT pathway, is related to tumor angiogenesis and metastasis. Therefore, we further explored the potential correlation between PRELP and FGF1. The negative correlation between PRELP and FGF1 protein expression was verified by western blot (Fig. 7A). Interestingly, we observed significantly enhanced colocalization between PRELP and FGF1 in cells overexpressing PRELP, as determined by immunofluorescence staining, suggesting a potential interaction (Fig. 7B). As expected, the immunoprecipitation results demonstrated that PRELP could interact with FGF1 (Fig. 7C). To further investigate the underlying mechanism of the decreased level of FGF1 after PRELP overexpression, we analyzed the influence of PRELP on the stability of the FGF1 protein. Intriguingly, compared with the vector group, the PRELP overexpression group exhibited accelerated degradation of FGF1 after CHX treatment (Fig. 7D). We also added the proteasome inhibitor MG132 to the cells stably overexpressing PRELP, and the results showed that the FGF1 protein was significantly increased after the addition of MG132 (Fig. 7E). The recombinant FGF1 protein was added to CRC cells in the PRELP group and vector group to further validate our findings. CCK-8 assay revealed that FGF1 partially rescued the inhibition of proliferation caused by PRELP (Fig. 7F). HUVECs were more likely to vascularize in the presence of FGF1 (Fig. 7G). Transwell assays also confirmed that the migration and invasion abilities of SW620 and RKO cells were restored by the recombinant FGF1 protein (Fig. 7H).Western blot analysis indicated that FGF1 could reactivate the PI3K/AKT/mTOR signaling pathway and promote the expression of VEGFA and EMT-related markers (Fig. 7I). Taken together, these findings indicated that PRELP could bind to FGF1 and reduce the stability of the FGF1 protein, accompanied by an increase in its degradation, which subsequently inactivated the PI3K/AKT/mTOR pathway, thereby resulting inreductions in tumor angiogenesis and metastasis.

PRELP bound to the FGF1 protein and affected its stability, thus inhibiting the PI3K/AKT/mTOR pathway. A Western blot analysis of PRELP associated with the FGF1 protein expression levels. B Immunofluorescence staining for PRELP and FGF1 in SW620 and RKO cells stably expressing PRELP. C Co-IP confirmed the interaction of PRELP with FGF1. D CHX (100 μg/mL) was added at different time points to explore the effect of PRELP on FGF1 protein stability. E CRC cells overexpressing PRELP were incubated with MG132 (10 μM) for 6 h and then the levels of FGF1 protein were detected by Western blot. F-I Cells stably expressing the vector and PRELP were treated with the recombinant FGF1protein (10 ng/ml) for 48 h and analyzed using a CCK-8 assay (F) tube formation assay (G) Transwell assay (H) and Western blot (I). * P < 0.05, ** P < 0.01, and *** P < 0.001
PRELP potentiated the antitumor efficacy of bevacizumab
Considering the negative regulatory effect of PRELP on VEGFA, we asked whether PRELP in combination with the VEGFA antagonist bevacizumab could improve the efficacy of antiangiogenic treatment. As expected, bevacizumab treatment further decreased the expression of VEGFA in cells overexpressing PRELP compared with that in the vector cells (Fig. 8A). Furthermore, we evaluated the in vivo efficacy of PRELP in combination with bevacizumab in a mouse xenograft model of SW620 cells stably overexpressing PRELP. We observed significantly decreased tumor growth in the PRELP group compared with the vector group after bevacizumab treatment. Notably, bevacizumab suppressed tumor growth both in the PRELP group and in the vector group (Fig. 8B–C). IHC of the xenograft tissue sections showed an obvious decrease in Ki-67, VEGFA, and CD31 expression with the addition of bevacizumab in the PRELP group in contrast to the vector group. Bevacizumab also reduced Ki-67, VEGFA, and CD31 expression in the PRELP group, as well as in the vector group (Fig. 8D). Taken together, these results suggested that PRELP could remarkably potentiate the efficacy of bevacizumab in vivo.

PRELP enhanced the efficacy of bevacizumab. A Bevacizumab (0.1 μg/mL) was added to SW620 and RKO cells cultured for 48 h, and VEGFA expression was analyzed using Western blot. B-D SW620 cells stably expressing the vector or PRELP were injected subcutaneously into one side of nude mice, which were randomly divided into two groups: those that received bevacizumab treatment or PBS. The tumors were collected after 21 days (B-C) and IHC analysis was performed (D). * P < 0.05, ** P < 0.01, and *** P < 0.001
Discussion
Recurrence and metastasis result in a poor prognosis for CRC patients. Tumor angiogenesis and the EMT are closely associated with tumor relapse and metastasis [27, 28]. The ECM plays an important role in the maintenance of the cellular structure and tissue homeostasis. Changes in ECM molecules lead to ECM degradation, which promotes angiogenesis and tumor cell migration and invasion [29]. As a significantly secreted proteoglycan in the ECM, PRELP has received increased attention for its function in tumors. The expression of PRELP is markedly reduced in hepatocellular carcinoma [14], bladder cancer [15], ovarian clear cell carcinoma [16], and retinoblastoma [17]. The absence of PRELP can reduce tumor cell adhesion [17]. In this study, we focused on the expression of PRELP in CRC and observed that PRELP was expressed at significantly lower levels in CRC tissues than in normal tissues. In addition, the level of PRELP expression in cancerous tissues was negatively correlated with the TNM stage and lymph node metastasis. PRELP expression was closely related to the prognosis of CRC patients. These findings indicated a tumor-suppressive role of PRELP in CRC.
We observed that PRELP inhibited CRC cell proliferation, halted the cell cycle, and promoted cell apoptosis. Moreover, PRELP also attenuated CRC invasion and migration by inhibiting the EMT and angiogenesis. Furthermore, PRELP significantly surppressed the stemness of SW620 and RKO cells. In addition, PRELP enhanced the efficacy of bevacizumab in CRC treatment. Thus, our current study provided evidence that PRELP could inhibit CRC progression and angiogenesis both in vivo and in vitro. Our study focused on the function of PRELP in CRC cells and endothelial cells. Because of the complexity of the ECM composition, it is necessary to explore the effects of PRELP on immune cells, fibroblasts, or other cells in future studies.
In recent years, researchers have reported several mechanisms of PRELP in tumors. Papadaki et al. reported that PRELP expression was significantly decreased in bladder cancer and inhibited cancer progression by suppressing the TGF-β and EGF pathways [30]. Histone acetylation-mediated HDACi alleviated PRELP gene repression in bladder cancer cells [15]. A recent study suggested that PRELP induced MHC class I and CCL5 expression and enhanced T cell immunogenicity in melanoma, thereby improving patient prognosis [18]. However, the mechanism of PRELP in CRC remains unknown. Our RNA-seq analysis revealed that DEGs in both SW620 and RKO cells were significantly enriched in the PI3K/AKT pathway. In vivo and in vitro experiments confirmed that PRELP inhibited CRC cell proliferation, angiogenesis, and metastasis by suppressing the PI3K/AKT/mTOR signaling pathway. We also used PI3K activators and inhibitors for rescue experiments. To the best of our knowledge, we are the first to report and experimentally confirm the mechanism by which PRELP inhibits CRC progression.
FGF1 is considered as a growth factor that regulates the proliferation, migration, and differentiation of target cells. FGF1 can promote the growth and migration of intestinal epithelial cells [31]. Activated fibroblasts can further secrete FGF1, leading to a vicious cycle of malignant phenotypes [32, 33]. Moreover, FGF1 is essential for vascular endothelial cell proliferation and is a potent angiogenic factor [34]. FGF1 also activates the PI3K/AKT pathway by binding to FGFR, which arouses our interest in whether PRELP is associated with FGF1. By performing co-IP, inhibitng protein synthesis and degradation, and rescue experiments, we confirmed that PRELP could interact with the FGF1 protein and promote its degradation, which resulted in the inhibition of the PI3K/AKT/mTOR signaling pathway. However, further studies are still needed to reveal the mechanism underlying the interaction between PRELP and FGF1. Studies have reported that FGF1 could be degraded via the ubiquitination pathway [35]. Therefore, we speculate that the degradation of FGF1 may be promoted through the ubiquitination-proteasome pathway, but relevant experiments still need to be conducted for verification in the future.
Angiogenesis has gained importance in tumor growth and metastasis. In recent years, growth factors, chemokines, and adhesion factors involved in tumor angiogenesis have been gradually reported. Targeted therapeutic studies based on these molecules have promoted the antiangiogenic treatment of tumors [36]. Endothelial and tumor cells, as well as the crosstalk between them, can be regulated by the PI3K/AKT pathway, which affects VEGF production by tumor cells and angiogenesis [37,38,39]. Our GO enrichment analysis of both SW620 and RKO cells suggested an association of PRELP with angiogenesis. Consistent with these findings, we observed more neovascularization in xenografts from the vector group than in those from the PRELP group in vivo. Further exploration revealed that PRELP could reduce VEGFA protein secretion by suppressing the PI3K/AKT signaling pathway. Bevacizumab, a humanized IgG monoclonal antibody targeting VEGFA, has been approved by the FDA as a first- and second-line VEGF-targeting agent for CRC [40]. By performing in vivo experiments, we showed that PRELP increased the efficacy of bevacizumab, which significantly inhibited tumor growth. These findings further support PRELP as a potential therapeutic target for CRC.
Conclusions
In conclusion, PRELP functions as a CRC suppressor. PRELP inhibits the PI3K/AKT/mTOR signaling pathway by interacting with FGF1 and accelerating its degradation, which suppresses the EMT and angiogenesis, ultimately inhibiting tumor progression. In addition, PRELP enhances the efficacy of bevacizumab. Our findings provide new insights into the function of PRELP in CRC and suggest a potential effective strategy for CRC treatment.
Data availability
The RNA-seq data of this article are available in the NCBI (https://www.ncbi.nlm.nih.gov/) with the dataset identifier PRJNA1102746. All data generated or analyzed during this study are included in this article and supplementary information files.
Abbreviations
- CRC:
-
Colorectal cancer
- EMT:
-
Epithelial-mesenchymal transition
- PRELP:
-
Proline/arginine-rich end and leucine-rich protein
- FGF1:
-
Fibroblast growth factor 1
- ECM:
-
Extracellular matrix
References
Siegel RL, Miller KD, Wagle NS, Jemal A (2023) Cancer statistics, 2023. CA Cancer J Clin 73(1):17–48. https://doi.org/10.3322/caac.21763
Arnold M, Sierra MS, Laversanne M, Soerjomataram I, Jemal A, Bray F (2017) Global patterns and trends in colorectal cancer incidence and mortality. Gut 66(4):683–691. https://doi.org/10.1136/gutjnl-2015-310912
Siegel RL, Miller KD, Goding Sauer A, Fedewa SA, Butterly LF, Anderson JC et al (2020) Colorectal cancer statistics, 2020. CA Cancer J Clin 70(3):145–164. https://doi.org/10.3322/caac.21601
Biller LH, Schrag D (2021) Diagnosis and treatment of metastatic colorectal cancer: a review. JAMA 325(7):669–685. https://doi.org/10.1001/jama.2021.0106
Stewart CL, Warner S, Ito K, Raoof M, Wu GX, Kessler J et al (2018) Cytoreduction for colorectal metastases: liver, lung, peritoneum, lymph, nodes, bone, brain. when does it palliate prolong survival and potentially cure? Curr Probl Surg 55(9):330–379. https://doi.org/10.1067/j.cpsurg.2018.08.004
Moserle L, Casanovas O (2013) Anti-angiogenesis and metastasis: a tumour and stromal cell alliance. J Intern Med 273(2):128–137. https://doi.org/10.1111/joim.12018
Lu W, Kang Y (2019) Epithelial-mesenchymal plasticity in cancer progression and metastasis. Dev Cell 49(3):361–374. https://doi.org/10.1016/j.devcel.2019.04.010
Zhou Q, Perakis SO, Ulz P, Mohan S, Riedl JM, Talakic E et al (2020) Cell-free DNA analysis reveals POLR1D-mediated resistance to bevacizumab in colorectal cancer. Genome Med. https://doi.org/10.1186/s13073-020-0719-6
Iozzo RV, Schaefer L (2015) Proteoglycan form and function: a comprehensive nomenclature of proteoglycans. Matrix Biol 42:11–55. https://doi.org/10.1016/j.matbio.2015.02.003
Bengtsson E, Mörgelin M, Sasaki T, Timpl R, Heinegård D, Aspberg A (2002) The leucine-rich repeat protein PRELP binds perlecan and collagens and may function as a basement membrane anchor. J Biol Chem 277(17):15061–15068. https://doi.org/10.1074/jbc.M108285200
Bengtsson E, Aspberg A, Heinegard D, Sommarin Y, Spillmann D (2000) The amino-terminal part of PRELP binds to heparin and heparan sulfate. J Biol Chem 275(52):40695–40702. https://doi.org/10.1074/jbc.M007917200
Grover J, Lee ER, Mounkes LC, Stewart CL, Roughley PJ (2007) The consequence of PRELP overexpression on skin. Matrix Biol 26(2):140–143. https://doi.org/10.1016/j.matbio.2006.10.005
Kosuge H, Nakakido M, Nagatoishi S, Fukuda T, Bando Y, Ohnuma SI et al (2021) Proteomic identification and validation of novel interactions of the putative tumor suppressor PRELP with membrane proteins including IGFI-R and p75NTR. J Biol Chem 296:100278. https://doi.org/10.1016/j.jbc.2021.100278
Hong R, Gu J, Niu G, Hu Z, Zhang X, Song T et al (2020) PRELP has prognostic value and regulates cell proliferation and migration in hepatocellular carcinoma. J Cancer 11(21):6376–6389. https://doi.org/10.7150/jca.46309
Shozu K, Kaneko S, Shinkai N, Dozen A, Kosuge H, Nakakido M et al (2022) Repression of the PRELP gene is relieved by histone deacetylase inhibitors through acetylation of histone H2B lysine 5 in bladder cancer. Clin Epigenet 14(1):147. https://doi.org/10.1186/s13148-022-01370-z
Dozen A, Shozu K, Shinkai N, Ikawa N, Aoyama R, Machino H et al (1999) Tumor suppressive role of the PRELP gene in ovarian clear cell carcinoma. J Pers Med 12(12):1999. https://doi.org/10.3390/jpm12121999
Hopkins J, Asada K, Leung A, Papadaki V, Davaapil H, Morrison M et al (2022) PRELP regulates Cell-Cell adhesion and EMT and inhibits retinoblastoma progression. Cancers (Basel) 14(19):4926. https://doi.org/10.3390/cancers14194926
Schäfer H, Subbarayan K, Massa C, Vaxevanis C, Mueller A, Seliger B (2023) Correlation of the tumor escape phenotype with loss of PRELP expression in melanoma. J Transl Med 21(1):643. https://doi.org/10.1186/s12967-023-04476-x
Ornitz DM, Itoh N (2022) New developments in the biology of fibroblast growth factors. WIREs Mech Dis 14(4):e1549. https://doi.org/10.1002/wsbm.1549
Ornitz DM, Itoh N (2015) The fibroblast growth factor signaling pathway. Wiley Interdiscip Rev Dev Biol 4(3):215–266. https://doi.org/10.1002/wdev.176
Chudzian J, Szlachcic A, Zakrzewska M, Czub M, Pustula M, Holak TA et al (2018) Specific antibody fragment ligand traps blocking FGF1 activity. Int J Mol Sci 19(9):2470. https://doi.org/10.3390/ijms19092470
Zhang J, Zhang J, Pang X, Chen Z, Zhang Z, Lei L et al (2021) MiR-205-5p suppresses angiogenesis in gastric cancer by downregulating the expression of VEGFA and FGF1. Exp Cell Res 404(2):112579. https://doi.org/10.1016/j.yexcr.2021.112579
Li P, Zhou D, Chen D, Cheng Y, Chen Y, Lin Z et al (2023) Tumor-secreted IFI35 promotes proliferation and cytotoxic activity of CD8+ T cells through PI3K/AKT/mTOR signaling pathway in colorectal cancer. J Biomed Sci 30(1):47. https://doi.org/10.1186/s12929-023-00930-6
Qadeer ZA, Valle-Garcia D, Hasson D, Sun Z, Cook A, Nguyen C et al (2019) ATRX in-frame fusion neuroblastoma is sensitive to EZH2 inhibition via modulation of neuronal gene signatures. Cancer Cell 36(5):512-527.e9. https://doi.org/10.1016/j.ccell.2019.09.002
Li L, Gong Y, Xu K, Chen W, Xia J, Cheng Z et al (2021) ZBTB28 induces autophagy by regulation of FIP200 and Bcl-XL facilitating cervical cancer cell apoptosis. J Exp Clin Cancer Res 40(1):150. https://doi.org/10.1186/s13046-021-01948-0
Abdulla M, Traiki TB, Vaali-Mohammed MA, El-Wetidy MS, Alhassan N, Al-Khayal K et al (2022) Targeting MUCL1 protein inhibits cell proliferation and EMT by deregulating β-catenin and increases irinotecan sensitivity in colorectal cancer. Int J Oncol 60(3):22. https://doi.org/10.3892/ijo.2022.5312
Guo Y, Guo Y, Chen C, Fan D, Wu X, Zhao L et al (2021) Circ3823 contributes to growth, metastasis and angiogenesis of colorectal cancer: involvement of miR-30c-5p/TCF7 axis. Mol Cancer 20(1):93. https://doi.org/10.1186/s12943-021-01372-0
Malki A, ElRuz RA, Gupta I, Allouch A, Vranic S, Al Moustafa AE (2020) Molecular mechanisms of colon cancer progression and metastasis: recent insights and advancements. Int J Mol Sci 22(1):130. https://doi.org/10.3390/ijms22010130
Su Z, Yang Z, Xu Y, Chen Y, Yu Q (2015) Apoptosis, autophagy, necroptosis, and cancer metastasis. Mol Cancer 14:48. https://doi.org/10.1186/s12943-015-0321-5
Papadaki V, Asada K, Watson JK, Tamura T, Leung A, Hopkins J et al (2020) Two secreted proteoglycans, activators of urothelial cell-cell adhesion, negatively contribute to bladder cancer initiation and progression. Cancers (Basel) 12(11):3362. https://doi.org/10.3390/cancers12113362
Dignass AU, Tsunekawa S, Podolsky DK (1994) Fibroblast growth factors modulate intestinal epithelial cell growth and migration. Gastroenterology 106(5):1254–1262. https://doi.org/10.1016/0016-5085(94)90017-5
Matsuki M, Adachi Y, Ozawa Y, Kimura T, Hoshi T, Okamoto K et al (2017) Targeting of tumor growth and angiogenesis underlies the enhanced antitumor activity of lenvatinib in combination with everolimus. Cancer Sci 108(4):763–771. https://doi.org/10.1111/cas.13169
Erdogan B, Webb DJ (2017) Cancer-associated fibroblasts modulate growth factor signaling and extracellular matrix remodeling to regulate tumor metastasis. Biochem Soc Trans 45(1):229–236. https://doi.org/10.1042/BST20160387
Li JP, Liu YJ, Zeng SH, Gao HJ, Chen YG, Zou X (2022) Identification of COX4I2 as a hypoxia-associated gene acting through FGF1 to promote EMT and angiogenesis in CRC. Cell Mol Biol Lett 27(1):76. https://doi.org/10.1186/s11658-022-00380-2
Wang M, Liu S, Wang H, Tang R, Chen Z (2020) Morphine post-conditioning-induced up-regulation of lncRNA TINCR protects cardiomyocytes from ischemia-reperfusion injury via inhibiting degradation and ubiquitination of FGF1. QJM 113(12):859–869. https://doi.org/10.1093/qjmed/hcaa088
Liu ZL, Chen HH, Zheng LL, Sun LP, Shi L (2023) Angiogenic signaling pathways and anti-angiogenic therapy for cancer. Sig Transduct Target Ther 8(1):198. https://doi.org/10.1038/s41392-023-01460-1
Soler A, Serra H, Pearce W, Angulo A, Guillermet-Guibert J, Friedman LS et al (2013) Inhibition of the p110α isoform of PI 3-kinase stimulates nonfunctional tumor angiogenesis. J Exp Med 210(10):1937–1945. https://doi.org/10.1084/jem.20121571
Xia C, Meng Q, Cao Z, Shi X, Jiang BH (2006) Regulation of angiogenesis and tumor growth by p110 alpha and AKT1 via VEGF expression. J Cell Physiol 209(1):56–66. https://doi.org/10.1002/jcp.20707
Jiang BH, Zheng JZ, Aoki M, Vogt PK (2000) Phosphatidylinositol 3-kinase signaling mediates angiogenesis and expression of vascular endothelial growth factor in endothelial cells. Proc Natl Acad Sci U S A 97(4):1749–1753. https://doi.org/10.1073/pnas.040560897
Xie YH, Chen YX, Fang JY (2020) Comprehensive review of targeted therapy for colorectal cancer. Sig Transduct Target Ther 5(1):22. https://doi.org/10.1038/s41392-020-0116-z
Acknowledgements
We would like to thank Dr. Jianwei Zhang at the First Affiliated Hospital of Chongqing Medical University for his kindly advice and excellent technical assistance.
Funding
This research was supported by the first affiliated hospital of Chongqing Medical University, Doctoral program of the first affiliated hospital of Chongqing Medical University (CYYY-BSYJSCXXM-202335).
Author information
Authors and Affiliations
Contributions
Xiaoqing Li and Zheng Jiang designed the study. Zhongxiang Jiang, Junfeng Li, and Kun Yang collected the clinical samples. Xiaoqing Li, Qianxi Deng, and Shuman Xu analyzed the data. Jin He, Zhihang Jiang, Fuqiang Liu, and Xiaoqing Li performed the animal experiments. Xiaoqing Li wrote the article. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
We declare that we do not have any commercial or affiliate interests that conflict with the submitted work.
Ethical approval
The Experimental Animal Management and Use Committee of Chongqing Medical University approved the experiment (approval number: IACUC-CQMU-2024–0269).
Consent for publication
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
10495_2024_2015_MOESM1_ESM.tif
Supplementary file1 (TIF 535733 KB)—Transwell raw images and histogram of western blot image grayscale value statistics.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Li, X., Jiang, Z., Li, J. et al. PRELP inhibits colorectal cancer progression by suppressing epithelial-mesenchymal transition and angiogenesis via the inactivation of the FGF1/PI3K/AKT pathway. Apoptosis (2024). https://doi.org/10.1007/s10495-024-02015-7
Accepted:
Published:
DOI: https://doi.org/10.1007/s10495-024-02015-7