
Abstract
Hypoxic pulmonary hypertension (HPH) is a pathophysiological syndrome in which pulmonary vascular pressure increases under hypoxic stimulation and there is an urgent need to develop emerging therapies for the treatment of HPH. LncRNA MIR210HG is a long non-coding RNA closely related to hypoxia and has been widely reported in a variety of tumor diseases. But its mechanism in hypoxic pulmonary hypertension is not clear. In this study, we identified for the first time the potential effect of MIR210HG on disease progression in HPH. Furthermore, we investigated the underlying mechanism through which elevated levels of MIR210HG promotes the transition from a contractile phenotype to a synthetic phenotype in PASMCs under hypoxia via activation of autophagy-dependent ferroptosis pathway. While overexpression of HIF-2α in PASMCs under hypoxia significantly reversed the phenotypic changes induced by MIR210HG knockdown. We further investigated the potential positive regulatory relationship between STAT3 and the transcription of MIR210HG in PASMCs under hypoxic conditions. In addition, we established both in vivo and in vitro models of HPH to validate the differential expression of specific markers associated with hypoxia. Our findings suggest a potential mechanism of LncRNA MIR210HG in the progression of HPH and offer potential targets for disease intervention and treatment.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Pulmonary hypertension (PH) is a clinical and pathophysiological syndrome characterized by elevated pulmonary artery pressure resulting from structural or functional changes in the pulmonary vessels [1]. In recent years, there has been a significant increase in the prevalence of PH across all age groups [2]. Hypoxic pulmonary hypertension (HPH) represents the third category of pulmonary hypertension. Prolonged exposure to hypoxia stimulates irreversible proliferation of pulmonary artery smooth muscle cells (PASMCs) and damages endothelial cells, leading to extensive pulmonary vascular lesions (such as intimal hyperplasia and thickening of the middle and outer membrane layers of muscular arteries, luminal stenosis and increased tension in non-muscular arteries, etc.), and eventually forming pulmonary hypertension [3].
Excessive proliferation of PASMCs serves as the underlying pathogenesis of PH, accompanied by the transition of PASMCs from a contractile phenotype to a synthetic phenotype [4, 5]. Normal PASMCs are highly differentiated elongated spindle-shaped cells, responsible for maintaining pulmonary vascular pressure through regulation of blood vessel contractility, but they lack the ability to proliferate and invade [6]. Nevertheless, when exposed to environmental factors such as hypoxia, PASMCs undergo transformation into undifferentiated rhombohedral synthetic cells, characterized by decreased expression of contractile markers (e.g., α-SMA, SMMHC), increased expression of synthetic cell markers (e.g., OPN, VIM), reduced cell contractile function and enhanced cell proliferation [7].
Ferroptosis is an iron-dependent programmed cell death process. When intracellular iron overload occurs, excessive Fe2+ catalyzes the production of lipid peroxides (LPO) and reactive oxygen species (ROS) through enzymatic or non-enzymatic reactions (Fenton reaction), and then accumulates cytotoxic aldehydes to induce cell death [8, 9]. Although the role of ferroptosis in PH remains controversial, it is evident that HPH pathogenesis involves an imbalance in iron homeostasis and an increase in intracellular LPO and ROS [10,11,12]. Autophagy represents one of the classical regulatory pathways associated with ferroptosis. In PASMCs of HPH, abnormal activation of autophagy provides ample nutrients for cell proliferation while exacerbating disease progression [12,13,14]. However, there have been no reports regarding whether autophagy regulates ferroptosis in PASMCs.
LncRNA MIR210HG is transcribed from the host gene MIR210HG, which is located on human chromosome 11p15.5. It is a long non-coding RNA closely related to hypoxia and has been widely reported in a variety of tumor diseases [15,16,17]. Previous studies have demonstrated that MIR210HG can interact with hypoxia-inducible factor family proteins through distinct mechanisms, leading to tumor cell proliferation, invasion, and epithelial-mesenchymal transition [16, 17]. However, the precise involvement of MIR210HG in regulating ferroptosis and its role in hypoxic pulmonary hypertension remains elusive. Signal transducer and activator of transcription 3 (STAT3) is a classical transcription factor, which participates in the transcription of multiple regulators of PH and induces the proliferation of PASMCs under hypoxia [18, 19].
In this study, we identified the potential effect of MIR210HG on phenotypic switching of PASMCs under hypoxia. Furthermore, we investigated the underlying mechanism through which MIR210HG affected disease progression in HPH by regulating PASMCs phenotypic switching. We further investigated the potential positive regulatory relationship of STAT3/MIR210HG/HIF-2α signaling axis in PASMCs under hypoxic conditions. In addition, we established both in vivo and in vitro models of HPH to validate the differential expression of specific markers associated with hypoxia.
Materials and methods
Cell culture and treatment
Primary human pulmonary arterial smooth muscle cells (hPASMCs) were purchased from the American Type Culture Collection (ATCC, VA, USA) and cultured in smooth muscle cell medium (ScienCell, CA, USA) supplemented with 10% fetal bovine serum (FBS), 1% SMC growth factor, and 1% penicillin/streptomycin. The cells were maintained in a normoxic environment at 21% O2, 5% CO2, and 37°C. To induce hypoxia, the cells were exposed to a culture condition consisting of 1% O2, 5% CO2, and 99% N2 for a duration of 24 to 48 h [20, 21]. Prior to treatment, all cells underwent a serum-free medium incubation period of 24 h.
Animal models
Twenty male Wistar rats, aged 6–8 weeks and weighing (210 ± 10 g), were procured from the Laboratory Animal Center of Anhui Medical University (Anhui, China). The rats were housed in cages under a 12-hour light/dark cycle with free access to water and food. They were randomly assigned into two groups: negative control (NC) group and HPH group, each consisting of 10 rats. The HPH model was induced by subjecting rats to an 8-hour daily exposure of air containing 10.0% oxygen, while the NC group was exposed to fresh air containing 21% oxygen [22]. After four weeks, hemodynamic parameters were measured to confirm the successful establishment of the HPH model. Pulmonary artery smooth muscle tissues from both groups were collected for subsequent research. This study protocol was approved by the Animal Ethics Committee of Anhui Medical University (Anhui, China).
Western blot
Cells and tissues were lysed using RIPA lysis buffer (Servicebio, Wuhan, China) supplemented with protease inhibitors for 30 min on ice, and the supernatant was collected. Total proteins were separated by electrophoresis on a 10% SDS-PAGE (Servicebio, Wuhan, China) and subsequently transferred onto polyvinylidene difluoride membranes. After blocking with 5% skim milk powder for 1 h, the membranes were incubated overnight at 4°C with primary antibodies. Subsequently, the membranes were incubated with horseradish peroxidase-conjugated IgG secondary antibody (Servicebio, Wuhan, China) for 1 h at room temperature [23, 24]. Western blot analysis was performed using a gel imaging system (Tanon, Shanghai, China). Primary antibodies used were as follows: STAT3 at 1:1000 (Bioworld, MN, USA); HIF-2α at 1:1000 (Bioworld, MN, USA); HIF-1α at 1:1000 (Bioworld, MN, USA); PTGS2 at 1:1000 (Bioworld, MN, USA); α-SMA at 1:1000 (Bioworld, MN, USA); OPN at 1:2000 (Abcam, Cambridge, UK); LC3B-II at 1:2000 (Abcam, Cambridge, UK); Beclin-1 at 1:1000 (Bioworld, MN, USA); β-actin at 1:5000 (Bioworld, MN, USA); GAPDH at 1:5000 (Bioworld, MN, USA).
Reverse transcription-polymerase chain reaction (RT-PCR)
Total RNA was isolated from cell samples using an RNA Quick Purification kit (Genuine, Henan, China). Complementary DNA (cDNA) templates were synthesized in accordance with the total RNA [25, 26]. Following the protocols provided by the assay kit and instrument manufacturers, the relative expression of MIR210HG was determined by real-time PCR using SYBR™ green master mix (Thermo, MA, USA) in a PRISM®7500 real-time PCR machine (ABI, CA, USA) and had been normalized by the expression of GAPDH. Nuclear and cytoplasmic RNA was isolated from the PASMCs under hypoxia using PARIS™ Kit (Thermo, MA, USA) to detect the subcellular localization of MIR210HG. GAPDH and U6 were used to normalize nuclear and cytoplasmic RNA, respectively. The primer pairs used were: MIR210HG-Forward: 5′-GCAGGCACAGGTGTGGTCATATC-3′ and MIR210HG-Reverse: 5′-AGGCAGGCTCAGCAGACAGG-3′; GAPDH-Forward: 5′-AGGTCGGTGTGAACGGATTTG-3′ and GAPDH-Reverse: 5′-TGTAGACCATGTAGTTGAGGTCA-3; U6-Forward: 5′-CTCGCTTCGGCAGCACA-3′ and U6-Reverse: 5′- AACGCTTCACGAATTTGCGT-3.
Cell transfection
GP-transfect-Mate (Genepharma, Shanghai, China) reagent was employed for the transfection of small interfering RNA (si-RNA, Genepharma, Shanghai, China) and pCMV-HIF-1α/HIF-2α/STAT3-3×FLAG-Neo (Fig. S1, Genepharma, Shanghai, China) into PASMCs in the logarithmic growth phase. The sequences for transfection were as follows: si-MIR210HG, sense: 5′-CAACACAGUUCACAAUAUATT-3′; anti-sense: 5′-UAUAUUGUGAACUGUGUUGTT-3′; si-HIF-2α, sense: 5′-CAGCAUCUUUGAUAGCAG-3′; anti-sense: 5′-ACUGCUAUCAAAGAUGCU-3′; si-STAT3, sense: 5′-GGGACCUGGUGUGAAUUAUTT-3′; anti-sense: 5′- AUAAUUCACACCAGGUCCCTT-3′. si-NC, sense: 5′-UUCUCCGAACGUGUCACGUTT-3′; anti-sense: 5′-ACGUGACACGUUCGGAGAATT-3′.
Dataset collection and bioinformatics analysis
Gene expression data from human pulmonary artery tissue are available on the GEO (https://www.ncbi.nlm.nih.gov/geo) dataset GSE236251 (platform: GPL11154), which contains 8 PAH patients (age 57.9 ± 4.1) and 8 control individuals (age 57.9 ± 2.7), and GSE188938 (platform: GPL23227), which contains 6 PAH patients (age 58.3 ± 8.8) and 4 control individuals (age 57.8 ± 8.5). HDOCK (http://hdock.phys.hust.edu.cn/) database was used to predict the binding ability of MIR210HG to HIF-2α protein, and LGscore > 1.5 and MaxSub > 0.1 were considered to have potential molecular interaction. JASPAR database (http://jaspar.genereg.net/) is used to predict the potential binding sites between STAT3 and MIR210HG promoter. The optimal promoter region was determined based on positive Strand and the highest Score as the selection criterion.
RNA pull-down assay
Biotin-labelled total MIR210HG probe and its antisense probe were synthesized from Genepharma (Genepharma, Shanghai, China), and PureBinding® RNA-protein pull-down Kit (Geneseed, Guangzhou, China) was used to perform the RNA pull-down assay. In brief, after hypoxic intervention of PASMCs for 24 h, the probes were incubated with cell lysates to form RNA-protein complexes. Subsequently, RNA-binding proteins were specifically captured using Streptavidin Magnetic Beads and HIF-2α enrichment levels were detected using Western Blot.
Chromatin immunoprecipitation (ChIP)
The ChIP assay was conducted using the ChIP assay kit (Beyotime, Shanghai, China). PASMCs were incubated in a medium containing 1% formaldehyde for 10 min to cross-link chromatin and proteins, and the cross-linking was terminated with glycine. After collecting and lysing the cells, the chromatin was ultrasonically fragmented into 200–500 bp fragments. One-tenth of the sonicated lysate served as the Input group, while the remaining lysates were incubated overnight at 4°C with STAT3 antibody and rabbit IgG separately. Protein A/G agarose was employed to enrich the antigen-antibody complex. The complex underwent delinking of crosslinking by heating at 65°C for 4 h after addition of 5 M NaCl. The products were digested with proteinase K, and the recovered DNA was utilized for PCR analysis. The primers designed to recognize the MIR210HG promoter sequence are provided below: MIR210HG-Forward: 5′- TGCAAAGATGCTTTCTCCCGAT − 3′ and MIR210HG -Reverse: 5′- GGCACCTTTTCTCAGCATCTGT − 3′.
Luciferase assay
The wild-type sequence containing the MIR210HG promoter was inserted into the pGL3-Basic vector (Fig. S1B, Genepharma, Shanghai, China) [22, 27]. Mutants were generated using a QuickChange® II mutagenesis kit (Agilent, CA, USA) and subsequently incorporated into the vector. GP-transfect-Mate (Genepharma, Shanghai, China) was employed for co-transfection the empty pGL3-basic vector or MIR210HG-WT plasmids or MIR210HG-MUT plasmids with the oe-STAT3 plasmid into PASMCs. Renilla luciferase was employed as the internal control gene and luciferase activity was assessed utilizing a Dual-Luciferase® assay kit (Promega, WI, USA).
Hematoxylin-eosin (HE) staining
According to the previous method [28], rat lung tissues were embedded in paraffin. After baking the paraffin sections at 65°C for 2 h, they were washed using xylene and alcohol. Sections were stained according to the HE staining method and the pulmonary arteries were captured through imaging with an orthotopic microscope (Leica, Wetzlar, Germany).
Immunofluorescence (IF) staining
The tissues and cells were fixed with 4% paraformaldehyde (Servicebio, Wuhan, China), followed by incubation with Triton-X100 (Servicebio, Wuhan, China) to permeabilize the membranes. After inactivating endogenous peroxidases, the samples were incubated overnight at 4°C with primary antibodies targeting PTGS2, OPN, and α-SMA. Afterward, they were exposed to Cy3 conjugated Goat Anti-Rabbit IgG (1:300, Servicebio, Wuhan, China) for 1 h at room temperature in the dark. To visualize nuclear morphology, counterstaining was performed using 4’,6-diamidino-2-phenylindole (DAPI, Servicebio, Wuhan, China). Pulmonary arterial images were acquired using an orthostatic fluorescence microscope while PASMCs were imaged utilizing a fluorescent inverted microscope (Olympus, Tokyo, Japan).
Cell counting Kit-8 (CCK-8) assay
Cell viability was detected using the CCK-8 kit (Servicebio, Wuhan, China). According to the reagent instructions, PASMCs (5*103/well) were seeded in 96-well plates and incubated in the CCk-8 working solution for 2 h in the dark after 24 h of hypoxic stimulation. The absorbance at 450 nm was measured using a microplate reader (Bio Tek, VT, USA).
ROS detection
The intracellular ROS level was detected by Reactive Oxygen Species Assay Kit (Servicebio, Wuhan, China). PASMCs were incubated in the dark at 37°C for 30 min using the DCFH-DA probe according to the manufacturer’s protocol. The fluorescence intensity was observed using a fluorescent inverted microscope (Olympus, Tokyo, Japan) an excitation wavelength of 488 nm and an emission wavelength of 525 nm.
LPO detection
The intracellular LPO level was detected by Cell Lipidperoxides Detection Kit (Dojindo, Kumamoto, Japan). PASMCs were incubated in the dark at 37°C for 30 min using the Liperfluo working solution according to the manufacturer’s protocol. The fluorescence intensity was observed using a live cell imager with an excitation wavelength of 488 nm and an emission wavelength of 525 nm.
GSH/GSSG assay
The intracellular reduced glutathione level was detected by GSH and GSSG Assay Kit (Beyotime, Shanghai, China). PASMCs were incubated in the dark at 25°C for 25 min using the total glutathione assay working solution according to the manufacturer’s protocol. The absorbance of the samples at a wavelength of 412 nm was measured using a microplate reader (Bio Tek, VT, USA).
Statistical analysis
All data were subjected to statistical analysis using GraphPad Prism 9.0 software. The non-parametric tests (Mann-Whitney test) was used to compare the significant differences between the two groups. A significance level of P < 0.05 was considered statistically significant.
Results
Hypoxia induces autophagy, ferroptosis and phenotype switching in pulmonary arteries of rats
In order to examine the programmed cell death patterns and phenotypic changes in the pulmonary vasculature of HPH rats, we established the hypoxia-induced rat model of PH. Histological analysis through HE staining revealed that, compared to the control group, the HPH group had significant thickening of the pulmonary arteries (Fig. 1A). Meanwhile, the hemodynamic measurements revealed a significant increase in mean pulmonary arterial pressure (mPAP), right ventricular systolic pressure (RVSP), and the ratio of right ventricular to left ventricular plus interventricular septum thickness (RV/(LV + S) within the HPH group, confirming the successful construction of the model (Fig. 1B–D). We collected total protein extracts from the pulmonary artery smooth muscle layers of the two rat groups. WB results showed that, in comparison to the control group, the autophagy markers Beclin-1 (Fig. 1F) and LC3-II (Fig. 1G) were significantly elevated in the HPH group. Additionally, ferroptosis markers PTGS2 (Fig. 1H) showed a significant increase, and the phenotype switching markers OPN (Fig. 1I) was upregulated, while α-SMA (Fig. 1J) was downregulated. Immunofluorescence was employed to further validate the expression levels of PTGS2 and OPN in the pulmonary artery smooth muscle layers of HPH rats (Fig. 1K-L). Compared to the control group, both proteins exhibited a significant increasing trend. In conclusion, we propose that in the pulmonary vasculature of HPH rats, the pulmonary artery smooth muscle layer undergoes autophagy, ferroptosis, and phenotype switching.

Autophagy, ferroptosis, and phenotypic switching in HPH rat model. (A) HE staining showed pulmonary artery thickening in the HPH rat model. (B–D) The levels of mPAP, RVSP, and RV/(LV + S) exhibited an increase in the HPH rat model. (E–J) The expression of Beclin-1, LC3B-II, PTGS2, OPN proteins was enhanced in a HPH rat model. While the expression of α-SMA was inhibited. (K, L) The expression of PTGS2 and OPN protein was enhanced in a hypoxia rat model with immunofluorescence staining analysis. mPAP: mean pulmonary arterial pressure, RVSP: right ventricular systolic pressure, RV/(LV + S): the ratio of right ventricle to left ventricle plus interventricular septum thickness. *P < 0.05, as compared with control, ***P < 0.001, as compared with control
MIR210HG regulates autophagy, ferroptosis and phenotype switching in PASMCs under hypoxia
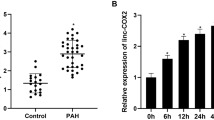
According to the previous method, we analyzed LncRNA expression profiles in two groups of hPASMCs (n = 3) subjected to normoxic or hypoxic stimulation [29]. A total of 154 differentially expressed genes (DEGs) were identified, consisting of 85 upregulated and 69 downregulated genes. The top 20 upregulated genes and the top 20 downregulated genes were visualized via a heatmap (Fig. 2A). Among the top 20 upregulated DEGs, LncRNA MIR210HG, which is closely associated with hypoxia, was identified as a pivotal molecule potentially involved in the regulation of HPH. Subsequently, we used GEO database to verify the expression of LncRNA MIR210HG (Fig. S2). Compared to controls, LncRNA MIR210HG showed significant elevation in lung tissue from PAH patients in both datasets. In addition, consistent with previous reports, PAH patients also had increased expression of ferroptosis markers PTGS2 or TFR1 (Fig. S2).

LncRNA MIR210HG regulates autophagy, ferroptosis, and phenotype switching in PASMCs under hypoxia. (A) The expression of the top 20 upregulated DEGs and the top 20 downregulated DEGs. (B) The expression of MIR210HG in PASMCs at 0, 4, 8, 12, 24, 36 h under hypoxia. (C) The expression of MIR210HG was increased under hypoxia, but was significantly inhibited after transfection with si-MIR210HG. (D–I) Si-MIR210HG inhibited the expression of Beclin-1, LC3B-II, PTGS2, OPN proteins in PASMCs under hypoxia. While the expression of α-SMA was enhanced. (J) Si-MIR210HG significantly increased reduced glutathione levels in PASMCs under hypoxia. (K–L) Si-MIR210HG reduced the fluorescence signal of LPO and ROS in PASMCs under hypoxia. (M) Si-MIR210HG enhanced the fluorescence signal of α-SMA in PASMCs under hypoxia. DEGs: differential expression genes, LPO: Lipid peroxides, ROS: reactive oxygen species. *P < 0.05, as compared with 0 h or si-NC, #P < 0.05, as compared with control, nsP>0.05, as compared with hypoxia, ***P < 0.001, as compared with si-NC
We detected MIR210HG expression under different hypoxia induction time, and found that its highest expression was induced after 24 h of hypoxia (Fig. 2B). Consequently, we determined this time point as the optimal duration for hypoxic treatment in cellular experiments. Subsequently, we constructed siRNA targeting MIR210HG and qPCR results demonstrated that siRNA significantly inhibited the expression of MIR210HG (Fig. 2C). Inhibition of MIR210HG under hypoxia led to a significant decrease in autophagy markers LC3-II, Beclin-1, and ferroptosis marker PTGS2 (Fig. 2E–G) compared with the control group. Simultaneously, after silencing MIR210HG, the intracellular GSH was significantly increased (Fig. 2J), and the levels of LPO and ROS were significantly decreased (Fig. 2K-L), which meant that ferroptosis was significantly inhibited in PASMCs. In addition, after MIR210HG inhibition, the expression of OPN was inhibited (Fig. 2H), the expression of SMA was enhanced (Fig. 2I), and the fluorescence signal of a-SMA was significantly enhanced compared with the control group (Fig. 2M), indicating that PAMSCs were reversed from synthetic phenotype to contractile type.
Ferroptosis regulate the phenotypic switching of PASMCs under hypoxia, which is mediated by MIR210HG
Subsequently, we validated whether the phenotype switching of PASMCs was regulated by ferroptosis. PASMCs were treated with the ferroptosis activator Erastin (MCE, USA) 10 μm for 24 h under normoxia according to previous research [30]. Compared to the control group, intracellular levels of LPO (Fig. 3A) and ROS (Fig. 3A) exhibited a significant increase, and there was an elevation in OPN (Fig. 3B), while α-SMA expression decreased (Fig. 3C). Meanwhile, under hypoxic conditions, we treated PASMCs with the ferroptosis inhibitor Ferrostain-1 (MCE, USA) at 5 μm for 24 h (Fig. S3). In comparison to the hypoxia group, Ferrostain-1 markedly suppressed intracellular LPO, ROS and OPN levels, and enhanced SMA expression (Fig. 3A–C). Rescue experiments further demonstrated that the Erastin could restore the decrease in OPN (Fig. 3D) and evidenced α-SMA fluorescence signals (Fig. 3E) caused by silencing MIR210HG under hypoxia. In conclusion, we suggest that MIR210HG can regulate the phenotypic switching of PASMCs under hypoxia through ferroptosis.

MIR210HG regulates the phenotypic switching of PASMCs under hypoxia through ferroptosis. (A) Erastin enhanced the fluorescence signal of LPO with ROS in PASMCs under normoxia, but ferrostatin-1 inhibited the fluorescence signal of LPO and ROS in PASMCs in PASMCs under hypoxia. (B-C) Erastin enhanced the OPN and inhibited the α-SMA in PASMCs under normoxia, but ferrostatin-1 inhibited the OPN and enhanced the α-SMA in PASMCs in PASMCs under hypoxia. (D) Erastin reversed the expression of OPN in PASMCs under hypoxia reduced by Si-MIR210HG. (E) Erastin reversed the fluorescence signal of α-SMA in PASMCs under hypoxia enhanced by Si-MIR210HG. LPO: Lipid peroxides, ROS: reactive oxygen species. *P < 0.05, as compared with control, #P < 0.05, as compared with hypoxia, &P < 0.05, as compared with si-MIR210HG
Ferroptosis regulates the phenotypic switching of PASMCs under hypoxia, which depends on the autophagy pathway
Previous literature indicates that ferroptosis is regulated by the autophagy pathway in various diseases. In accordance with prior research, we inhibited autophagy in PASMCs using the autophagy inhibitor 3-Methyladenine (3-MA, MCE, USA) at 5 mM. Compared to the control group, inhibition of hypoxia-induced autophagy in PASMCs resulted in a significant reduction in autophagy makers Beclin-1 (Fig. 4A), LC3-II (Fig. 4B). Despite an increase in PTGS2 (Fig. S4), the ferroptosis marker TFR1 was markedly suppressed (Fig. 4F). Additionally, intracellular levels of LPO (Fig. 4H) and ROS (Fig. 4I) were significantly reduced, indicating that the ferroptosis in PASMCs remained inhibited. Furthermore, after inhibiting autophagy of PASMCs under hypoxia, OPN decreased (Fig. 4D) while SMA increased (Fig. 4E) and the fluorescence signal of α-SMA was notably enhanced (Fig. 4J), marking the cells by synthetic phenotype reply for the contractive phenotype.

Autophagy regulates ferroptosis, and phenotype switching in PASMCs under hypoxia. (A–F) 3-MA inhibited the expression of Beclin-1, LC3B-II, TFR1, OPN proteins in PASMCs under hypoxia. While the expression of α-SMA was enhanced. (G) 3-MA significantly increased reduced glutathione levels in PASMCs under hypoxia. (H-I) 3-MA reduced the fluorescence signal of LPO and ROS in PASMCs under hypoxia. (J) 3-MA enhanced the fluorescence signal of α-SMA in PASMCs under hypoxia. 3-MA: 3-Methyladenine, LPO: Lipid peroxides, ROS: reactive oxygen species. *P < 0.05, as compared with control, **P < 0.01, as compared with control
MIR210HG regulates ferroptosis and phenotype switching of PASMCs under hypoxia through autophagy
The potential regulatory mechanism of MIR210HG on phenotypic switching of PASMCs was further verified by rescue experiments. Based on our previous research, the autophagy activator rapamycin 50 μm (Rapa, MCE, USA) was used to induce autophagy of PASMCs. Compared with si-MIR210HG group, Rapa inducing autophagy flux LC3-II reactivated (Fig. 5C), and make the intracellular PTGS2 (Fig. 5D), ROS (Fig. 5A), LPO (Fig. 5A) significantly increased. In addition, Rapa also reversed the decrease in OPN (Fig. 5E), and an enhancement of the fluorescence signal of α-SMA (Fig. 5F) caused by inhibition of MIR210HG. Our results confirmed that MIR210HG could promote phenotypic switching of PASMCs under hypoxia through autophagy-dependent ferroptosis pathway.

MIR210HG regulates the ferroptosis and phenotypic switching of PASMCs under hypoxia through autophagy. (A) Rapa enhanced the fluorescence signal of LPO and ROS in PASMCs reduced by Si-MIR210HG. (B–E) Rapa enhanced the expression of LC3B-II, PTGS2, OPN proteins in PASMCs under hypoxia reduced by Si-MIR210HG. (F) Rapa inhibited the fluorescence signal of α-SMA in PASMCs under hypoxia enhanced by Si-MIR210HG.Rapa: Rapamycin, LPO: Lipid peroxides, ROS: reactive oxygen species. *P < 0.05, as compared with control, #P < 0.05, as compared with hypoxia, &P < 0.05, as compared with hypoxia + si-MIR210HG
MIR210HG regulates autophagy, ferroptosis, and phenotypic switching in PASMCs under hypoxia through HIF-2α
Multiple HIF family proteins are involved in the regulation of ferroptosis, and MIR210HG has been reported to bind to HIF to promote tumor cell proliferation. Therefore, we examined the expression of HIF-1α and HIF-2α in PASMCs under different treatments. Consistent with previous reports, hypoxia significantly increased the expression of HIF-1α (Fig. S5B) and HIF-2α (Fig. 6A) in PASMCs, while both were significantly inhibited when MIR210HG was silenced in the cells. Subsequently, by separating nuclear and cytoplasmic RNA, we found that MIR210HG mainly localized in the cytoplasm of PASMCs (Fig. 6B). The secondary structure analysis using the HDOCK database revealed a good binding propensity (LGscore = 3.098, MaxSub = 0.270) between MIR210HG and HIF-2α (Fig. S5A). RNA pulldown further confirmed that MIR210HG significantly enriched HIF-2α signaling in PASMCs under hypoxia compared to the sense group (Fig. 6C-D).

MIR210HG regulates the autophagy, ferroptosis and phenotypic switching of PASMCs under hypoxia through HIF-2α. (A) Si-MIR210HG inhibited the expression of HIF-2α in PASMCs under hypoxia. (B) Nuclear and cytoplasmic expression levels of LncRNA MIR210HG, GAPDH mRNA, and U6 in PASMCs under hypoxia. (C-D) HIF-2α binding to MIR210HG was detected by RNA pulldown. (E) Oe-HIF-2α promoted the expression of HIF-2α in hypoxic PASMCs, but si-HIF-2α inhibited it. (F–I) Oe-HIF-2α enhanced the expression of LC3B-II, PTGS2, OPN proteins in PASMCs under hypoxia decreased by si-MIR210HG. (J) Oe-HIF-2α enhanced the fluorescence signal of LPO and ROS in PASMCs under hypoxia inhibited by si-MIR210HG. (K) Oe-HIF-2α inhibited the fluorescence signal of α-SMA expression in PASMCs under hypoxia increased by si-MIR210HG. *P < 0.05, as compared with control, #P < 0.05, as compared with hypoxia or hypoxia + si-MIR210HG
We generated overexpression plasmids and si-RNA targeting HIF-2α, and Western blot analysis demonstrated significant activation or inhibition of HIF-2α expression (Fig. 6E). Consistent with silencing of MIR210HG, knockdown of HIF-2α resulted in decreased protein levels of LC3-II, PTGS2 and OPN in PASMCs under hypoxia. Conversely, overexpression of HIF-2α significantly activated the expression of these markers that were suppressed by MIR210HG knockdown (Fig. 6F–I). Silencing HIF-2α also attenuated the fluorescence signals of LPO and ROS in PASMCs under hypoxia, while activation of HIF-2α reversed the ferroptosis level of PASMCs inhibited by silencing MIR210HG (Fig. 6J). However, overexpression of HIF-1α failed to restore ferroptosis level in PASMCs (Fig. S5D). In addition, immunofluorescence results showed that activation of HIF-2α in PASMCs under hypoxia also restored the α-SMA fluorescence signal expression enhanced by MIR210HG inhibition (Fig. 6K). These results confirmed that MIR210HG activated autophagy, ferroptosis and phenotype switching in PASMCs under hypoxia by directly targeting and regulating HIF-2α.
STAT3 regulates MIR210HG, autophagy, ferroptosis and phenotype switching in PASMCs under hypoxia
The activation of STAT3 plays a pivotal role in promoting the abnormal proliferation of PASMCs in HPH. Our experiments also confirmed that STAT3 was significantly increased in PASMCs under hypoxia (Fig. 7A). Then, siRNA targeting STAT3 was transfected into PASMCs and Western blot results showed that si-STAT3 significantly inhibited the expression of STAT3 (Fig. 7C). In addition, silencing STAT3 inhibited the autophagy flux LC3-II (Fig. 7D), decreased the ferroptosis markers PTGS2 (Fig. 7E) and LPO (Fig. 7G), and enhanced the contractile phenotype marker α-SMA (Fig. 7F). These results provide preliminary evidence that STAT3 regulates autophagy, ferroptosis and phenotype switching in PASMCs under hypoxia. Subsequently, we constructed a STAT3 overexpression plasmid and used Western blot to demonstrate that oe-STAT3 could promote STAT3 activation (Fig. 7H). The qPCR results showed that MIR210HG in cells was changed with the change of STAT3 levels (Fig. 7I), implying that STAT3 is able to regulate the expression of MIR210HG in PASMCs under hypoxia.

STAT3 regulates MIR210HG, autophagy, ferroptosis, and phenotype switching in PASMCs under hypoxia. (A) The expression of STAT3 was increased in PASMCs under hypoxia. (B–F) Si-STAT3 inhibited the expression of STAT3, LC3B-II, PTGS2 proteins in PASMCs under hypoxia. While the expression of α-SMA was enhanced. (G) Si-STAT3 reduced the fluorescence signal of LPO in PASMCs under hypoxia. (H) oe-STAT3 enhanced the expression of STAT3 in PASMCs under hypoxia. (I) oe-STAT3 promoted the expression of MIR210HG in hypoxic PASMCs, but si-STAT3 inhibited it. LPO: Lipid peroxides. *P < 0.05, as compared with control or si-NC or oe-NC
STAT3 regulates autophagy, ferroptosis and phenotype switching in PASMCs under hypoxia by promoting MIR210HG transcription
STAT3 is a classic transcription factors promote variety of gene transcription in PASMCs. To verify whether STAT3 was involved in the transcriptional regulation of MIR210HG, we used Jaspar database to predict whether there was a potential binding site between STAT3 and MIR210HG promoter region, and found a short promoter fragment at -1610–1600 bp upstream of MIR210HG gene. We built primers for this fragment, and ChIP assay confirmed that the potential promoter fragments specifically pulled down by STAT3 under hypoxia were significantly higher than that under normoxia (Fig. 8A). Furthermore, we extracted 2000 bp upstream of the MIR210HG gene and inserted it into a plasmid to construct a wild-type MIR210HG promoter vector (Fig. 8B). Meanwhile the potential transcription factor binding site (TFBS) was mutated to construct the mutant MIR210HG promoter vector (Fig. 8C). Luciferase reporter assay showed that the luciferase activity was significantly inhibited after mutation of this site (Fig. 8D), which further confirmed that STAT3 was the key signal promoting MIR210HG transcription. Finally, we found that overexpression of STAT3 in PASMCs under hypoxia significantly increased the levels of LC3-II, PTGS2, OPN, LPO and ROS (Fig. 8F–J), while slightly reducing the fluorescence intensity of α-SMA (Fig. 8I) compared to the control group. However, these observations were effectively reversed upon inhibition of MIR210HG in PAMSCs. All in all, our results confirmed that STAT3 could promote autophagy, ferroptosis and phenotype switching in PASMCs under hypoxia by regulating the transcription of MIR210HG.

STAT3 promotes MIR210HG transcription and regulates the autophagy, ferroptosis and phenotypic switching of PASMCs under hypoxia through MIR210HG. (A) The degree of STAT3 binding to MIR210HG promoter was higher in PASMCs under hypoxia than under normoxia. (B-C) Wild-type and mutant MIR210HG promoter sequences were constructed, and the binding site of MIR210HG and STAT3 were predicted. (D) STAT3 binds the wild-type MIR210HG promoter and promotes transcription, but mutating this binding site inhibits this process. (E–H) Si-MIR210HG reduced the expression of LC3B-II, PTGS2, OPN proteins in PASMCs under hypoxia increased by oe-STAT3. (I) Si-MIR210HG enhanced the fluorescence signal of α-SMA in PASMCs under hypoxia inhibited by oe-STAT3. (J) Si-MIR210HG inhibited the fluorescence signal of LPO and ROS in PASMCs enhanced by oe-STAT3. LPO: Lipid peroxides, ROS: reactive oxygen species. *P < 0.05, as compared with control, #P < 0.05, as compared with hypoxia, &P < 0.05, as compared with hypoxia + oe-STAT3
Discussion
In the advanced stage of chronic lung disease, long-term hypoxia often leads to PH. However, vasodilators used to treat pulmonary hypertension often aggravate the oxygenation and ventilation conditions, and there is an urgent need to develop emerging therapies for the treatment of HPH [31]. Abnormal proliferation of PASMCs is the basis for the pathogenesis of pulmonary vascular hypertrophy in HPH, while the transition of PASMCs from a contractile phenotype to a synthetic phenotype is an early event in this phenomenon. In recent years, more and more research focused on the molecular pathway to regulate this process, include HIF-1α pathway [32], MAPK pathway [33], PI3k/ATK pathway [34], etc. Although the relationship between ferroptosis and phenotypic switching is not clear, but hypoxia can increase the level of nicotinamide adenine dinucleotide phosphate oxidase in PASMCs, leading to intracellular ROS accumulation and mitochondrial dysfunction. Consequently, these alterations activate changes in markers associated with phenotypic switching [5, 35]. In addition, the literature has shown that ferroptosis can induce the transition of vascular smooth muscle cells to a synthetic phenotype and neointima formation by promoting the accumulation of ROS in carotid artery ligation mice model [36]. In our research, we have firstly elucidated the pivotal role of the ferroptosis pathway in driving the phenotypic transition of PASMCs towards a synthetic state. When we activated ferroptosis of PASMCs under normoxia or inhibited ferroptosis of PASMCs under hypoxia, there was a corresponding increase or decrease of the synthetic marker OPN and a corresponding inhibition or reactivation of the contractile marker α-SMA.
As an emerging mode of programmed cell death, the investigation into ferroptosis in PH is still in its nascent stage. At present, the role of ferroptosis in PH is still controversial. Although activation of ferroptosis has been reported to inhibit PASMCs proliferation [30], most studies have identified ferroptosis as a key contributor to pulmonary hypertension [37,38,39]. On the one hand, the levels of MDA, ROS and labile iron pool were increased and mitochondrial damage in MCT-induced PAECs [38], on the other hand, ferrostatin-1 can significantly alleviate right ventricular fibrosis, remodeling and dysfunction in HPH rats [37]. We suggest that hypoxia can reduce the threshold of ferroptosis induction in PASMCs by disrupting the balance between anti-ferroptosis and pro-ferroptosis factors [12]. Consistent with previous studies, we also observed the accumulation of LPO and activation of ferroptosis in PASMCs under hypoxia [30]. However, treatment with a ferroptosis inhibitor, but not an activator, ameliorated hypoxia-induced pulmonary hypertension by reducing the sensitivity of PASMCs to ferroptosis (Fig. S3). In addition, we further found that ferroptosis promote pulmonary hypertension is dependent on the activation of autophagy pathway. When hypoxia induces excessive autophagy in PSMACs cells, a large amount of ferritin degradation leads to intracellular iron accumulation. On one hand, excessive levels of free iron can directly trigger ferroptosis through the Fenton reaction. On the other hand, it can impair mitochondrial autophagy, resulting in the accumulation of ROS and LPO, ultimately aggravating ferroptosis [40]. In our study, we observed changes in intracellular ROS, LPO, ferroptosis markers TFR1 and PTGS2, as well as phenotypic switching markers by modulating autophagy flux in PASMCs, demonstrating for the first time the crucial involvement of the autophagy-dependent ferroptosis pathway in HPH.
Long non-coding RNA (LncRNA) plays a crucial role in regulating gene expression through various dimensions such as epigenetics, transcriptional regulation, and post-transcriptional regulation [41]. Recently, the involvement of LncRNA in HPH has garnered increasing attention as a potential therapeutic target. In our study, based on LncRNA microarray expression profile changes in HPH, we identified LncRNA MIR210HG as an important regulatory signal in HPH. Interestingly, the expression of MIR210HG under hypoxia was closely related to HIF family proteins, and there was a potential interaction between MIR210HG and HIF. In ovarian cancer cells, MIR210HG directly bound to HIF-1α and inhibited the degradation of HIF-1α to promote cancer progression [42]. In cervical cancer cells, MIR210HG and HIF-1α affected the level of cell glycolysis through a positive feedback loop, contributing to the occurrence of cervical cancer [43]. A recent study also revealed abnormal elevation of MIR210HG in pulmonary artery endothelial cells under hypoxia, with its transcript level regulated by KMT2E-AS1/H3K4me3 [44]. This study further found a positive feedback loop that the transcription of KMT2E-AS1 was also regulated by HIF-2α, and KMT2E-AS1 increased the expression of HIF-2α by increasing HIF transcription and controlling HIF degradation. It is worth noting that HIF family proteins play a role in regulating ferroptosis progression in cells. Previous studies have suggested that HIF-1α induces ferroptosis resistance in cells, while HIF-2α serves as a crucial signal for activating ferroptosis. In addition, HIF-2α plays a significant role in the phenotypic switching of PASMCs in the early stage of pulmonary hypertension [45, 46]. This study provides the pioneering evidence elucidating the regulatory function of MIR210HG in PASMCs under hypoxia. Our results confirmed that MIR210HG also increased HIF-2α in cells by binding to HIF-2α and inhibiting its degradation, thereby activating autophagy-dependent ferroptosis and phenotype switching regulation of PASMCs in the early stage of HPH. STAT3 is a classical transcriptional regulator, and extensive research has focused on its potential therapeutic efficacy as a signaling pathway in HPH. Currently, many drugs have been identified to effectively ameliorate pulmonary hypertension by specifically targeting the inhibition of STAT3 phosphorylation to modulate the JAK-STAT pathway [47, 48]. In addition, STAT3 is also involved in the transcription of some key genes in HPH. It is reported that STAT3 could bind to the miR-146b promoter to induce pulmonary vascular remodeling in HPH via the QKI-STAT3-miR-146b pathway [49]. Our study provides additional evidence supporting the involvement of STAT3 in the transcriptional regulation of HPH. We first proved that MIR210HG is under the regulation of STAT3, which directly binds to the promoter region of MIR210HG to promote its transcription and increase its expression level of MIR210HG in PASMCs under hypoxia.
Our study still has some limitations. Firstly, our findings have been substantiated through rigorous external experimental validation. However, comprehensive investigations involving human subjects are need to provide further evidence for targeted therapy. In addition, we take hypoxia as the sole cause of this disease, and more realistic environmental factors need to be simulated to clarify the pathogenesis of PAH.
Conclusion
In this study, we demonstrated a novel mechanism by which MIR210HG regulates the progression of HPH. Specifically, the transcription of MIR210HG is regulated by STAT3 and activated MIR210HG promotes the transition from a contractile phenotype to a synthetic phenotype in PASMCs under hypoxia by interacting with HIF-2α to increase intracellular levels of HIF-2α and then activating the autophagy-dependent ferroptosis pathway. Our findings provide new insights into the pathogenesis of HPH and offer potential targets for disease intervention and treatment.
Data availability
No datasets were generated or analysed during the current study.
References
Humbert M, Kovacs G, Hoeper MM et al (2023) 2022 ESC/ERS guidelines for the diagnosis and treatment of pulmonary hypertension. Eur Respiratory J Jan 61(1):2200879. https://doi.org/10.1183/13993003.00879-2022
Hoeper MM, Humbert M, Souza R et al (2016) A global view of pulmonary hypertension. Lancet Respiratory Med Apr 4(4):306–322. https://doi.org/10.1016/s2213-2600(15)00543-3
Ruffenach G, Hong J, Vaillancourt M, Medzikovic L, Eghbali M (2020) Pulmonary hypertension secondary to pulmonary fibrosis: clinical data, histopathology and molecular insights. Respir Res Nov 18(1):303. https://doi.org/10.1186/s12931-020-01570-2
Owens GK, Kumar MS, Wamhoff BR (2004) Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol Rev Jul 84(3):767–801. https://doi.org/10.1152/physrev.00041.2003
Ma B, Cao Y, Qin J, Chen Z, Hu G, Li Q (2023) Pulmonary artery smooth muscle cell phenotypic switching: a key event in the early stage of pulmonary artery hypertension. Drug Discov Today May 28(5):103559. https://doi.org/10.1016/j.drudis.2023.103559
Wang G, Jacquet L, Karamariti E, Xu Q (2015) Origin and differentiation of vascular smooth muscle cells. J Physiol Jul 15(14):3013–3030. https://doi.org/10.1113/jp270033
Frid MG, Aldashev AA, Dempsey EC, Stenmark KR (1997) Smooth muscle cells isolated from discrete compartments of the mature vascular media exhibit unique phenotypes and distinct growth capabilities. Circ Res Dec 81(6):940–952. https://doi.org/10.1161/01.res.81.6.940
Zhang H, Zhou S, Sun M et al (2022) Ferroptosis of endothelial cells in Vascular diseases. Nutrients Oct 26(21):4506. https://doi.org/10.3390/nu14214506
Dixon SJ, Stockwell BR (2014) The role of iron and reactive oxygen species in cell death. Nat Chem Biol Jan 10(1):9–17. https://doi.org/10.1038/nchembio.1416
Xie SS, Deng Y, Guo SL et al (2022) Endothelial cell ferroptosis mediates monocrotaline-induced pulmonary hypertension in rats by modulating NLRP3 inflammasome activation. Sci Rep Feb 23(1):3056. https://doi.org/10.1038/s41598-022-06848-7
Wong CM, Preston IR, Hill NS, Suzuki YJ (2012) Iron chelation inhibits the development of pulmonary vascular remodeling. Free Radic Biol Med Nov 1(9):1738–1747. https://doi.org/10.1016/j.freeradbiomed.2012.08.576
Wang E, Zhou S, Zeng D, Wang R (2023) Molecular regulation and therapeutic implications of cell death in pulmonary hypertension. Cell Death Discov Jul 12(1):239. https://doi.org/10.1038/s41420-023-01535-6
Chen YB (2019) Autophagy and its role in pulmonary hypertension. Aging Clin Exp Res Aug 31(8):1027–1033. https://doi.org/10.1007/s40520-018-1063-1
Antonioli M, Di Rienzo M, Piacentini M, Fimia GM (2017) Emerging mechanisms in initiating and terminating Autophagy. Trends Biochem Sci Jan 42(1):28–41. https://doi.org/10.1016/j.tibs.2016.09.008
Zou F, Zhang ZH, Zou SS et al (2023) LncRNA MIR210HG promotes the proliferation, migration, and invasion of lung cancer cells by inhibiting the transcription of SH3GL3. Kaohsiung J Med Sci Dec 39(12):1166–1177. https://doi.org/10.1002/kjm2.12775
Saigusa H, Mimura I, Kurata Y, Tanaka T, Nangaku M (2023) Hypoxia-inducible lncRNA MIR210HG promotes HIF1alpha expression by inhibiting mir-93-5p in renal tubular cells. FEBS J Aug 290(16):4040–4056. https://doi.org/10.1111/febs.16794
Hu XL, Huang XT, Zhang JN et al (2022) Long noncoding RNA MIR210HG is induced by hypoxia-inducible factor 1alpha and promotes cervical cancer progression. Am J Cancer Res 12(6):2783–2797
Darnell JE Jr., Kerr IM, Stark GR (1994) Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. J Article Rev Sci Jun 3(5164):1415–1421. https://doi.org/10.1126/science.8197455
Hu WP, Xie L, Hao SY et al (2022) Protective effects of progesterone on pulmonary artery smooth muscle cells stimulated with interleukin 6 via blocking the shuttling and transcriptional function of STAT3. Article. Int Immunopharmacol Jan 102:108379. https://doi.org/10.1016/j.intimp.2021.108379
Liu YY, Zhang WY, Zhang ML et al (2022) DNA-PKcs participated in hypoxic pulmonary hypertension. Respir Res Sep 16(1):246. https://doi.org/10.1186/s12931-022-02171-x
Ding X, Zhou S, Li M et al (2017) Upregulation of SRF is Associated with Hypoxic Pulmonary hypertension by promoting viability of smooth muscle cells via increasing expression of Bcl-2. J Cell Biochem Sep 118(9):2731–2738. https://doi.org/10.1002/jcb.25922
Xia X, Huang L, Zhou S et al (2023) Hypoxia-induced long non-coding RNA plasmacytoma variant translocation 1 upregulation aggravates pulmonary arterial smooth muscle cell proliferation by regulating autophagy via miR-186/Srf/Ctgf and miR-26b/Ctgf signaling pathways. Int J Cardiol Jan 1 370:368–377. https://doi.org/10.1016/j.ijcard.2022.09.060
Zhou S, Sun L, Cao C et al (2018) Hypoxia-induced microRNA-26b inhibition contributes to hypoxic pulmonary hypertension via CTGF. J Cell Biochem Feb 119(2):1942–1952. https://doi.org/10.1002/jcb.26355
Zhou S, Liu Y, Li M et al (2018) Combined effects of PVT1 and MiR-146a single nucleotide polymorphism on the lung function of smokers with chronic obstructive Pulmonary Disease. Int J Biol Sci 14(10):1153–1162. https://doi.org/10.7150/ijbs.25420
Sun L, Xu A, Li M et al (2021) Effect of methylation status of lncRNA-MALAT1 and MicroRNA-146a on pulmonary function and expression level of COX2 in patients with Chronic Obstructive Pulmonary Disease. Front Cell Dev Biol 9:667624. https://doi.org/10.3389/fcell.2021.667624
Zhu K, Zhou S, Xu A et al (2020) Microbiota Imbalance contributes to COPD Deterioration by enhancing IL-17a production via miR-122 and miR-30a. Mol Ther Nucleic Acids Dec 4:22:520–529. https://doi.org/10.1016/j.omtn.2020.09.017
Zhou S, Zhu K, Du Y et al (2020) Estrogen administration reduces the risk of pulmonary arterial hypertension by modulating the miR-133a signaling pathways in rats. Gene Ther Apr 27(3–4):113–126. https://doi.org/10.1038/s41434-019-0103-6
Wang C, Liu Y, Zhang W et al (2022) circ-BPTF serves as a mir-486-5p sponge to regulate CEMIP and promotes hypoxic pulmonary arterial smooth muscle cell proliferation in COPD. Acta Biochim Biophys Sin (Shanghai) Dec 25(3):438–448. https://doi.org/10.3724/abbs.2022178
Wang E, Wang Y, Zhou S et al (2022) Identification of three hub genes related to the prognosis of idiopathic pulmonary fibrosis using bioinformatics analysis. Int J Med Sci 19(9):1417–1429. https://doi.org/10.7150/ijms.73305
Hu P, Xu Y, Jiang Y et al (2022) The mechanism of the imbalance between proliferation and ferroptosis in pulmonary artery smooth muscle cells based on the activation of SLC7A11. Article. Eur J Pharmacol Aug 5:928:175093. https://doi.org/10.1016/j.ejphar.2022.175093
Hoeper MM, Barberà JA, Channick RN et al (2009) Diagnosis, assessment, and treatment of non-pulmonary arterial hypertension pulmonary hypertension. J Am Coll Cardiol Jun 30(1 Suppl):85–96. https://doi.org/10.1016/j.jacc.2009.04.008
Ahmad A, Ahmad S, Malcolm KC et al (2013) Differential regulation of pulmonary vascular cell growth by hypoxia-inducible transcription factor-1alpha and hypoxia-inducible transcription factor-2alpha. Article. Am J Respir Cell Mol Biol Jul 49(1):78–85. https://doi.org/10.1165/rcmb.2012-0107OC
Yan L, Gao H, Li C, Han X, Qi X (2017) Effect of miR-23a on anoxia-induced phenotypic transformation of smooth muscle cells of rat pulmonary arteries and regulatory mechanism. Article. Oncol Lett Jan 13(1):89–98. https://doi.org/10.3892/ol.2016.5440
Wang GS, Qian GS, Zhou DS, Zhao JQ (2005) JAK-STAT signaling pathway in pulmonary arterial smooth muscle cells is activated by hypoxia. Article. Cell Biol Int Jul 29(7):598–603. https://doi.org/10.1016/j.cellbi.2005.03.014
Nowak WN, Deng J, Ruan XZ, Xu Q (2017) Reactive Oxygen species Generation and Atherosclerosis. Article. Arterioscler Thromb Vasc Biol May 37(5):41–52. https://doi.org/10.1161/ATVBAHA.117.309228
Zhang S, Bei Y, Huang Y et al (2022) Induction of ferroptosis promotes vascular smooth muscle cell phenotypic switching and aggravates neointimal hyperplasia in mice. Article. Mol Med Oct 3(1):121. https://doi.org/10.1186/s10020-022-00549-7
Song J, Chen Y, Chen Y et al (2023) Ferrostatin-1 blunts right ventricular hypertrophy and dysfunction in pulmonary arterial hypertension by suppressing the HMOX1/GSH signaling. J Cardiovasc Transl Res Aug 21. https://doi.org/10.1007/s12265-023-10423-4
Liao J, Xie SS, Deng Y et al (2023) PRDX6-mediated pulmonary artery endothelial cell ferroptosis contributes to monocrotaline-induced pulmonary hypertension. Article. Microvasc Res Mar 146:104471. https://doi.org/10.1016/j.mvr.2022.104471
Vogel NT, Annis J, Prisco S et al (2023) Ferroptosis promotes pulmonary hypertension. Preprint bioRxiv Dec 2. https://doi.org/10.1101/2023.01.19.524721
Lee S, Hwang N, Seok BG, Lee S, Lee SJ, Chung SW (2023) Autophagy mediates an amplification loop during ferroptosis. Rev Cell Death Disease Jul 25(7):464. https://doi.org/10.1038/s41419-023-05978-8
Kopp F, Mendell JT (2018) Functional Classification and Experimental Dissection of Long Noncoding RNAs. Cell. Jan 25. ;172(3):393–407. https://doi.org/10.1016/j.cell.2018.01.011
Liu P, Huang H, Qi X et al (2021) Hypoxia-Induced LncRNA-MIR210HG promotes Cancer Progression by inhibiting HIF-1α degradation in Ovarian Cancer. Front Oncol 11:701488. https://doi.org/10.3389/fonc.2021.701488
Hu XL, Huang XT, Zhang JN et al (2022) Long noncoding RNA MIR210HG is induced by hypoxia-inducible factor 1α and promotes cervical cancer progression. Am J Cancer Res 12(6):2783–2797
Tai YY, Yu Q, Tang Y et al (2024) Allele-specific control of rodent and human lncRNA KMT2E-AS1 promotes hypoxic endothelial pathology in pulmonary hypertension. Sci Transl Med Jan 10(729):eadd2029. https://doi.org/10.1126/scitranslmed.add2029
Zhou X, Zheng Y, Sun W et al (2021) D-mannose alleviates osteoarthritis progression by inhibiting chondrocyte ferroptosis in a HIF-2α-dependent manner. Cell Prolif Nov 54(11):e13134. https://doi.org/10.1111/cpr.13134
Chan XY, Volkova E, Eoh J et al (2021) HIF2A gain-of-function mutation modulates the stiffness of smooth muscle cells and compromises vascular mechanics. iScience Apr 23(4):102246. https://doi.org/10.1016/j.isci.2021.102246
Yerabolu D, Weiss A, Kojonazarov B et al (2021) Targeting Jak-Stat Signaling in Experimental Pulmonary Hypertension. Am J Respir Cell Mol Biol Jan 64(1):100–114. https://doi.org/10.1165/rcmb.2019-0431OC
Fu M, Luo F, Wang E et al (2021) Magnolol attenuates right ventricular hypertrophy and fibrosis in Hypoxia-Induced Pulmonary arterial hypertensive rats through inhibition of the JAK2/STAT3 signaling pathway. Front Pharmacol 12:755077. https://doi.org/10.3389/fphar.2021.755077
Huang H, Lin D, Hu L et al (2023) RNA binding protein quaking promotes hypoxia-induced smooth muscle reprogramming in Pulmonary Hypertension. Article. Am J Respir Cell Mol Biol Aug 69(2):159–171. https://doi.org/10.1165/rcmb.2022-0349OC
Acknowledgements
We sincerely thank Dr. Haijie Wang for her assistance during the preparation of this manuscript.
Funding
This research was supported by the fund for Natural Science Foundation of China (No.81970051), A sub-project of the Anhui Medical University National first-class undergraduate specialty construction program (clinical medicine), Young Jianghuai famous medical training project, Research Fund project of Anhui Medical University (2023xkj144), Excellent physician Training program of Anhui Medical University, Research Fund of Anhui Institute of translational medicine (2023zhyx-C40), Construction projects of key disciplines in Hefei (Occupational medicine), Health Research Project of Anhui Province (AHWJ2023A30009) and the Applied Medical Research Project of Hefei Health Commission (Hwk2021zd008, Hwk2022zd013).
Author information
Authors and Affiliations
Contributions
EW and BZ analyzed the data and wrote the first draft. LH and SZ designed the study, DZ and RW revised the submission. PL and RH directed the statistical analyses of the data. All authors have read and agreed to the submitted version of the manuscript.
Corresponding authors
Ethics declarations
Ethics committee approval
All animal experiments in this study were approved by the Animal Experimental Ethics Committee of Anhui Medical University and were performed in strict accordance with the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health.
Competing interests
The authors have declared that no competing interest exists.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Wang, E., Zhang, B., Huang, L. et al. LncRNA MIR210HG promotes phenotype switching of pulmonary arterial smooth muscle cells through autophagy-dependent ferroptosis pathway. Apoptosis 29, 1648–1662 (2024). https://doi.org/10.1007/s10495-024-01963-4
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10495-024-01963-4