
Abstract
It is essential to further characterize liver injury aimed at developing novel therapeutic approaches. This study investigated the mechanistic basis of genipin against carbon tetrachloride (CCl4)-triggered acute liver injury concerning ferroptosis, a novel discovered modality of regulated cell death. All experiments were performed using hepatotoxic models upon CCl4 exposure in mice and human hepatocytes in vitro. Immunohistochemistry, immunoblotting, molecular docking, RNA-sequencing and ultra-high-performance liquid chromatography-tandem mass spectrometry (UHPLC-MS/MS) were conducted. CCl4 intoxication was manifested with lipid peroxidation-dictated ferroptotic cell death, together with changes in a cascade of ferroptosis-associated events and several regulatory pathways. Both the administration of genipin and ferrostatin-1 (Fer-1) significantly prevented this hepatotoxicity in response to CCl4 intoxication via upregulating GPX4 and xCT (i.e., critical regulators of ferroptosis). RNA-sequencing unraveled that arachidonic acid metabolism was considerably influenced upon genipin treatment. Accordingly, genipin treatment attenuated arachidonate 15-lipoxygenase (ALOX15)-launched lipid peroxidation in terms of UHPLC-MS/MS analysis and inflammation. In vitro, genipin supplementation rescued erastin-induced hepatocellular inviability and lipid ROS accumulation. The siRNA knockdown of GPX4 partially abrogated the protective effects of genipin on erastin-induced cytotoxicity, whereas the cytotoxicity was less severe in the presence of diminished ALOX15 expression in L-O2 cells. In conclusion, our findings uncovered that genipin treatment protects against CCl4-triggered acute liver injury by abrogating hepatocyte ferroptosis, wherein the pharmacological modification of dysregulated GPX4 and ALOX15-launched lipid peroxidation was responsible for underlying medicinal effects as molecular basis.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
It has been documented a considerable mortality rate (around 10%) of hospitalized patients due to drug-induced liver injury [1]. Furthermore, acute liver injury partially accounts for the advent, development and progression of various liver diseases which can result in terminal organ failure. Frequent and recurrent liver injury instigates fibrosis, cirrhosis and sometimes the aggressive phenotype as hepatocellular carcinoma. The main reasons of liver injury comprise hepatotoxicant, drug overdose, alcoholism, inappropriate self-medication as well as viral hepatitis infection [2]. Taking into consideration the heavy burden on health systems and public concern pertinent to hepatotoxicant/drug intoxication, increasing attention and many efforts have been dedicated to elucidate the molecular basis of acute liver injury, if available, to develop potential therapeutic approaches.
As for carbon tetrachloride (CCl4), this hepatotoxic substance has been widely used to establish well-known experimental model regarding acute liver injury, indicative of necrotic alterations involving different zones in the hepatic lobes [3]. Of note, toxicants stemming from CCl4 metabolite exhibit high affinity to lipids, and in turn remove the H● from the membranous unsaturated fatty acids which are responsible for the subsequent chain process of lipid peroxidation to elicit hepatocellular damages. This toxicant injury model is suited to assess the underlying mechanism in relation to hepatic damages and to identify potential hepatoprotective/anti-hepatotoxic effects of bio-synthetic agents, as well as ingredients derived from natural products [4]. Moreover, CCl4-triggered liver injury is characterized by similar morphological and biochemical shifts coincided with human liver disorders [5]. Since CCl4 intoxication to liver accounts for multiple detrimental biological processes, such as oxidative stress, endoplasmic reticulum stress, apoptosis, autophagy and ferroptosis, emerging evidence implicates the potentials to counteract these CCl4-triggered deleterious effects by identifying therapeutic interventions on the basis of natural/herbal sources [6].
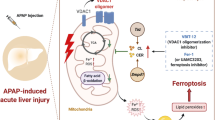
Ferroptosis, as an iron catalysis-mediated novel form of regulated cell death, is appreciated as excessive production of lipid peroxides. Recent researches demonstrate that a wide spectrum of polyunsaturated fatty acids (PUFAs), including arachidonic acid (AA), may participate in the ferroptotic pathways [7]. The peroxidation of lipids encompassing PUFA chains leads to further accumulation of lethal lipid reactive oxygen species (ROS) [8]. Accordingly, lipid peroxidation accounts for the final executor of ferroptosis, which is facilitated by arachidonate lipoxygenase (ALOX)-mediated oxidative response in an enzymatically reactive fashion [9]. Conversely, intracellular glutathione peroxidase 4 (GPX4) is capable of preventing fatal chain reaction through converting resultant 15-hydroperoxyeicosatetraenoyl acid (15-HpETE) to reduced 15-hydroxyeicosatetraenoyl acid (15-HETE) [10]. Until now, a growing body of literature has explored the role of ferroptosis in the context of various liver diseases [11,12,13]. However, there is scant data concerning the impact of ALOX-launched lipid peroxidation and the combination of divergent ferroptotic pathways in CCl4-triggered acute liver injury [1, 14, 15].
Despite a variety of bio-synthetic medications have been used to treat liver injury/hepatic dysfunction, their usage are restrained due to limited therapeutic effects, intolerance to prescription and unexpected side effects such as renal and cardiac toxicity in clinical settings. Therefore, aforesaid drawbacks dramatically arouse scientific endeavor to identify hepatoprotective alternatives among the traditional Chinese medicine (TCM). Actually, TCM has been long and broadly used in Oriental countries as efficious complementary therapeutics [16]. Genipin (Fig. 1A) is the aglycone derived from geniposide, the most abundant iridoid glucoside constituent of Gardenia jasminoides Ellis, getting conversion through intestinal bacteria-produced enzyme β-D-glycosidase [17]. Thereafter, genipin is absorbed via the intestine and transported to liver across the portal vein. Our previous report implicated that genipin protects against CCl4-triggered hepatotoxicity, which is linked to the activation of autophagy [18].

Genipin protects against acute liver injury subjected to CCl4 intoxication in vivo. (A) The chemical structure of genipin. (B) Schematic illustration of the experimental protocols in detail. (C, D) Alterations of macroscopic (scale bar: 10 mm) and microscopic (scale bar: 100 μm) appearance in terms of H&E staining. (E, F) Measurement of serum ALT/AST levels. (G) Evaluation of histopathological changes in terms of HAI score. ###p < 0.001 vs. control group, +++p < 0.001 vs. CCl4 group, &&&p < 0.001 vs. CCl4 group
Taking into consideration the concept that ferroptosis can determine cell fate in addition to the contribution of hepatocellular iron storage, the current study elaborated on the role of GPX4 pathway and ALOX15-launched lipid peroxidation for acute liver injury in the context of ferroptosis. Herein, our findings unveiled that a cascade of ferroptosis-associated events, including mitochondrial morphology alterations, accumulation of lipid ROS, dysregulation of key regulator GPX4 and accumulation of lipid peroxidation mediated by ALOX15, is observed in mice under CCl4 exposure. Further, we sought to clarify the medicinal effects of genipin against CCl4 intoxication. To validate genipin as a potential therapeutic agent, we utilized RNA-sequencing, molecular docking and ultra-high-performance liquid chromatography-tandem mass spectrometry (UHPLC-MS/MS) methods; further analyses showed that the administration of genipin and ferrostatin-1 (Fer-1) effectively mitigates toxicant-induced ferroptosis via upregulating GPX4 and suppressing ALOX15-launched lipid peroxidation. Finally, the shifts in the targeted ferroptotic pathways upon erastin (a pharmacological reagent to induce ferroptosis) exposure were reversed with genipin treatment in vitro, which is then confirmed using transfection knockdown of GPX4 or ALOX15 [19].
Materials and methods
We showed additionally available materials and experimental protocols in detail referring to the supplementary files (Supplementary File).
Animal toxicant injury models, treatment and experimental design
All animal studies were approved by the Institutional Animal Care and Use Committee at Tianjin Medical University General Hospital (IRB2021-DWFL-142). Mice were purchased from the National Institutes for Food and Drug Control (Beijing, China). The mice were allowed for one week of acclimation period to minimize environmental differences. The mice were housed in a pathogen-free room maintained under specific conditions: a temperature of 23 ± 2 °C and relative humidity of 50 ± 10% with a 12 h light-dark cycle, together with water and food ad libitum. Male C57BL/6 mice aged 6–8 weeks and weighed 20–22 g, unless otherwise indicated, were employed. As for the acute liver injury model, the mice were subjected to a dose of 2 ml/kg combining CCl4 (50%) and olive oil (50%) via an intraperitoneal (i.p.) injection. The control group was indicative of an i.p. injection for the same value of olive oil as the CCl4 group. The liver and blood samples were collected 48 h afterward. As for the pharmacological modification, an intravenous injection of genipin or saline (vehicle) was performed via the tail vein 2 h prior to CCl4 exposure. In this experiment, we chose a 48 h time point and an optimally effective dose of 2.5 mg/kg genipin for the whole protocols coincided with previous reports (Fig. 1B) [18, 20]. Fer-1 was dissolved in saline (1 mg/kg) along with an i.p. injection to mice for once 1 h prior to CCl4 challenge aimed at elaborating on the medicinal effects of genipin on CCl4-triggerd liver injury in the context of ferroptosis. The mice were randomly divided into six groups (per each group 3 ~ 6 mice): (1) vehicle-treated normal control (control); (2) vehicle-treated CCl4 challenge (CCl4); (3) 1 mg/kg Fer-1-treated CCl4 challenge (CCl4 + Fer-1); (4) 1 mg/kg Fer-1-treated (Fer-1); (5) 2.5 mg/kg genipin-treated CCl4 challenge (CCl4 + genipin); (6) 2.5 mg/kg genipin-treated (genipin).
Cell culture and siRNA knockdown experiment
Normal human hepatocyte L-O2, purchased from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China), were incubated in Dulbecco’s Modified Eagle Medium encompassing fetal bovine serum (10%), penicillin (100 IU/ml) and streptomycin (100 mg/ml) in 37 °C with 5% CO2. In this experiment, L-O2 hepatocytes were transfected with non-targeting control siRNA (NC siRNA, 80 pmol/ml), siRNA directed against ALOX15 (80 pmol/ml) or GPX4 (80 pmol/ml) for 48 h by employing Lipofectamine 3000 in terms of the manufacturer’s instruction.
Statistical analysis
All data were depicted in the manner regarding mean ± standard deviation (SD). The overall significance of data was compared in terms of one-way analysis of variance, and statistical differences among several groups were considered significance at p < 0.05 with the Bonferroni correction in the case of multiple comparisons. We used GraphPad Prism 8.0.1 (Graph Pad Software, Inc. San Diego, CA, U.S.) to analyze data in the current study.
Results
The execution of ferroptosis in response to CCl4-triggered acute liver injury
First, we sought to clarify the role of ferroptosis in the context of CCl4-triggerd acute liver injury. In alignment with our previous report, the 48 h time point was of choice coincided with the most pronounced histological damages in the mice liver [18]. Regarding the macroscopic appearance and H&E staining findings, the destruction and hepatic damages due to CCl4 exposure were consistently alleviated upon Fer-1 treatment, designated as a specific ferroptosis inhibitor by capturing/eliminating lipophilic radicals (Fig. 1C, D). Simultaneously, the aggravation concerning histopathological scores in the injurious mice liver was significantly mitigated by Fer-1 supplementation (Fig. 1G). Moreover, we found similar medicinal effects regarding genipin treatment, mirrored as diminishment of hepatocellular necrosis, hepatic architecture loss as well as inflammatory cell infiltration. Accordingly, our findings unraveled that Fer-1 supplementation considerably reverses increases in ALT/AST concentration in response to CCl4 challenge as compared with the control group (Fig. 1E, F).
As for TEM analysis, we showed that the mitochondria are smaller in size alongside reduced cristae; the outer membrane is torn in mice liver due to CCl4 exposure (Fig. 2A). In addition, our findings uncovered that, via TUNEL assay, both administration of Fer-1 and genipin remarkably repress hepatocyte death (TUNEL (+) cells) in the injurious mice livers (Fig. 2B, C). In line with therapeutic potentials against liver damages, genipin and Fer-1 treatment dramatically restored CCl4-triggered mitochondrial destruction relative to untreated samples. Altogether, these data strongly supported the hypothesis that execution of ferroptosis is responsible for CCl4-triggered acute liver injury.

Genipin represses hepatocyte death in vivo. (A) Analysis of TEM for mitochondrial morphology of ferroptotic cells in the liver tissue (red arrow; scale bar: 500 nm). (B) Measurement of dead hepatocytes in terms of TUNEL staining in the mice livers (scale bar: 100 μm). Representative images were present. (C) Quantification of TUNEL (+) cells was present. Data were expressed as mean ± SD. ###p < 0.001 vs. control group, +++p < 0.001 vs. CCl4 group, &&&p < 0.001 vs. CCl4 group
Genipin represses CCl4-triggered ferroptotic process in vivo
As for typically biochemical features in the context of ferroptosis, for instance, Fe2+ accumulation, extensive ROS generation and resulting lipid peroxides, we demonstrated that genipin effectively represses these detrimental activities. The expression levels of 4-HNE and MDA, suggestive of toxicant-induced lipid peroxidation serving as critical executors for ferroptosis, were significantly diminished in response to the genipin treatment (Fig. 3A, D). Using specific 581/591 C11-BODIPY fluorescent probe, a marked production of lipid ROS was observed in the liver tissue challenged by CCl4. By contrast, no lipid ROS accumulation was found in the control group. Additionally, both supplementation of genipin and Fer-1 significantly reverse increases of lipid ROS/hepatic ROS levels compared to CCl4 challenged group (Fig. 3B, C).

Genipin represses CCl4-triggered ferroptotic process in vivo. (A) Detection of 4-HNE protein adducts expression in fixed liver tissue sections (scale bar: 100 μm). (B) Detection of accumulated lipid ROS in terms of 581/591 C11-BODIPY fluorescent probe (scale bar: 100 μm). (C) Measurement of hepatocellular ROS in the liver tissue. (D) Assessment of the content of MDA in the liver tissue. (E) Evaluation of hepatic Fe2+ levels. (F) Determination of Ptgs2 gene expression in terms of real-time PCR analysis. Data were expressed as mean ± SD. ##p < 0.01, ###p < 0.001 vs. control group, +p < 0.05, +++p < 0.001 vs. CCl4 group, &p < 0.05, &&&p < 0.001 vs. CCl4 group
Dysregulated iron metabolism contributes to the induction of ferroptosis among a wide spectrum of liver diseases [21,22,23]. Our findings showed that Fe2+ levels increase in the liver of CCl4-triggered mice, whereas Fer-1 and genipin ameliorate these deleterious iron overload (Fig. 3E). Moreover, a validated biomarker of ferroptotic pathway Ptgs2 gene expression was diminished upon genipin treatment relative to the injurious liver models (Fig. 3F) [24, 25].
Genipin upregulates hepatic expression of GPX4 and xCT in vivo
It has been built that GPX4 and xCT (a multipass transmembrane protein encoded by the solute carrier family 7 member 11 gene) serve as pivotal regulators of ferroptosis, thus we sought to determine the pertubations in protein expression levels. Our findings revealed that CCl4 exposure downregulates the protein levels of GPX4 around 26.5% relative to the control group, but genipin rescued these decreases by upregulating around three fold over the injured group (Fig. 4A, B). Furthermore, the expressed xCT levels significantly decreased to 24.3% in CCl4-intoxicated livers compared to the controls. In the CCl4 + genipin group, the expressed GPX4 levels increased closely to two fold relative to CCl4 exposure group. Of note, Fer-1 also restored the decreases of GPX4/xCT due to CCl4 intoxication. Taken together, these data argued a critical role of hepatocyte ferroptosis in the context of CCl4-triggered acute liver injury and the therapeutic potentials to suppress ferroptosis with genipin in vivo.

Genipin regulates hepatic expression of GPX4/xCT and several key mediators in relation to ferroptotic process in vivo. (A) Determination of protein levels for xCT and GPX4 in the liver tissue subjects to CCl4 challenge. (B) Quantification of the band intensities for xCT and GPX4 relative to β-actin in terms of image J software. (C) Determination of hepatic protein levels for FTH1, p53 and p21 upon genipin treatment in the context of toxicant-induced ferroptosis. (D) Quantification of the band intensities for FTH1, p53 and p21 relative to GAPDH. (E) Determination of FTH1 and FTL gene expression in terms of real-time PCR analysis. Data were expressed as mean ± SD. **p < 0.01 vs. control group, ***p < 0.001 vs. control group, +++p < 0.001 vs. CCl4 group, &&p < 0.01 vs. CCl4 group, &&&p < 0.001 vs. CCl4 group
Genipin affects several key mediators in relation to ferroptotic process
Accumulating evidence suggests that multiple mediators responsible for the ferroptotic process concerning distinct pillars. For instance, the role of p53-p21 pathway in the context of ferroptosis appears to be complicated. It is suggested that p53-KR abrogates cystine import and in consequence leads to ferroptotic cell death, whereas p53-p21 activation may also potentiate glutathione (GSH) recycle along with decrease in cellular GSH export/consumption [26, 27]. The ferritin heavy chain 1 (FTH1) is associated with sensitivity to ferroptosis, indicative of silencing FTH1 towards aggravated erastin-induced ferroptosis [28]. As shown in Fig. 4C, D, the positive ferroptosis mediators p53-p21 were significantly upregulated in CCl4-intoxicated livers, whereas the expression of FTH1 proteins decreased in the CCl4 exposure group relative to the control group. The gene expression of hepatic FTH1 and ferritin light chain (FTL) also decreased in the CCl4 exposure group (Fig. 4E). Particularly, genipin treatment effectively restored these alterations. Thereafter, we sought to measure inflammation and lipid peroxidation in the context of ferroptosis in vivo. As for F4/80 staining positive macrophages in response to CCl4 exposure, the increases were mitigated by supplementation with genipin and Fer-1 (Figure S1A, B). Hepatic p21 and ALOX15 gene expressions were upregulated in CCl4-triggered mice, whereas administration of genipin and Fer-1 inhibited these increases (Figure S1C, D). Given CCl4 can result in liver damages through extensive inflammatory response and a potential link between necroinflammation and ferroptosis, we detected the serum levels of typical cytokines. As shown in Table S1, CCl4 significantly stimulated the production of IL-1β, IL-6 and TNF-α in the circulation relative to the control group, whereas both genipin and Fer-1 supplementation suppressed these proinflammatory cytokines.
Genipin inhibits erastin-induced hepatocyte ferroptosis in vitro
In an attempt to elucidate the role and molecular basis pertaining to ferroptosis in the context of liver injury in vitro, we applied several pharmacological inhibitors including Fer-1, Z-VAD-FMK and Nec-1 to identify subroutines of regulated cell death upon erastin treatment. Our findings implicated that both genipin and Fer-1 ameliorate erastin-induced hepatocyte death to some extent (Fig. 5A). In contrast, neither Z-VAD-FMK nor Nec-1 rescued cell inviability. The accumulation of LDH and MDA was also mitigated in response to genipin and Fer-1 treatment compared with the erastin group (Fig. 5B, C). As for cellular ROS, the DCFH-DA probe demonstrated that genipin remarkably reduces the severity of lipid peroxidation elicited by erastin in L-O2 cells (Fig. 5D). As for altered morphology via TEM evaluation, genipin restored mitochondrial shrinkage induced by erastin (Fig. 5E). As for iron homeostasis, the excessive cellular and mitochondrial Fe2+ accumulation due to erastin challenge were observed in terms of higher FerroOrange and Mito-FerroGreen signals relative to the controls, but these changes were significantly attenuated upon genipin and Fer-1 administration (Fig. 5F, G).

Genipin inhibits erastin-induced hepatocyte ferroptosis in vitro. (A) Assessment of the viability of L-O2 cells challenged with erastin and divergent pharmacological inhibitors including Z-VAD-FMK, Nec-1, Fer-1 and genipin in terms of CCK-8. (B, C) Evaluation of the accumulation of LDH and MDA in L-O2 cells. (D) Quantification of cellular ROS content in terms of DCFH-DA probe (scale bar: 200 μm). (E) Analysis of TEM pertinent to mitochondrial morphology of ferroptotic L-O2 cells (scale bar: 500 nm). (F, G) Detection of cellular and mitochondrial Fe2+ culmination in terms of FerroOrange probe and Mito-FerroGreen probe of cultured L-O2 cells (scale bar: 200 μm). ###p < 0.001 vs. control group, ++p < 0.01 vs. erastin group, +++p < 0.001 vs. erastin group, &&p < 0.01, &&&p < 0.001 vs. erastin group
Modifying GPX4 partially accounts for hepatoprotective effects of genipin
Given the essential role of GPX4 to counteract the generation of lipid ROS in the presence of Fe2+ (catalytically active iron), a molecular docking analysis of GPX4 with genipin was implemented (Fig. 6A). Using this approach, genipin was considered to embrace relatively high binding affinity with amino acid residue Val-125 of GPX4 to orchestrate an intermolecular hydrogen bond (XP docking score: -3.422).

Genipin exerts hepatoprotective effects by modifying GPX4. (A) Molecular docking analysis of GPX4 with genipin. (B) Determination of protein levels of GPX4 in L-O2 cells transfected with NC siRNA or GPX4 siRNA. (C) Assessment of the viability of L-O2 cells transfected with NC siRNA or GPX4 siRNA. (D) Evaluation of the accumulation of LDH in L-O2 cells transfected with NC siRNA or GPX4 siRNA. (E, G) Detection of cellular ROS content in L-O2 cells transfected with NC siRNA or GPX4 siRNA in terms of DCFH-DA probe (scale bar: 200 μm). (F, H) Detection of TEM pertinent to mitochondrial morphology of ferroptotic L-O2 cells (scale bar: 500 nm). ###p < 0.001 vs. control group, αp < 0.05 vs. GPX4 siRNA group, αααp < 0.001 vs. GPX4 siRNA group
Next, the medicinal effects of genipin were validated by siRNA targeting GPX4. As shown in Fig. 6B, we selected si-GPX4#2 with high transfection efficiency for all further experiments. Accordingly, GPX4 knockdown remarkably aggravated erastin-induced cytotoxicity, and genipin administration failed to rescue cell cytotoxicity/damages in this circumstance (Fig. 6C, D). As for DCFH-DA and TEM analyses, these findings also supported the pivotal role of GPX4 contributing to the therapeutic potential of genipin (Fig. 6E-H). Collectively, all in vitro data verified that genipin has hepatoprotective effects by modifying GPX4 pathway to counteract toxicant-induced ferroptosis.
Inhibiting ALOX15-launched lipid peroxidation partially accounts for hepatoprotective effects of genipin
To further elaborate on the mechanistic basis by which genipin exerted hepatoprotective effects, we analyzed an RNA-sequencing dataset for CCl4-triggered and genipin treatment mice. The KEGG pathway analysis showed that several biological processes, including fatty acid degradation, biosynthesis of unsaturated fatty acids, fatty acid elongation as well as AA metabolism were enriched within the top 10 pathways (Fig. 7A, B). Actually, those pathways are in close relation to ferroptosis execution, since n-6 PUFAs like AA in phospholipid membranes can react with ROS [29]. Given the significance of lipoxygenase, known as a non-heme, iron-containing enzyme capable of catalyzing the PUFAs deoxygenation, we further detected ALOX15-launched lipid peroxidation. By using UHPLC-MS/MS, we found that several oxylipins associated with ALOX15 catabolism including AA, 15-HpETE and 15-HETE are upregulated in CCl4-intoxicated livers, and increases in these lipid peroxides were reversed with genipin treatment (Fig. 7C). Furthermore, we conducted in vitro experiments to validate the role of ALOX15 to facilitate ferroptotic process. In terms of ALOX15 protein silencing by siRNA approach (Fig. 7D), we showed that ALOX15 knockdown (using si-ALOX15#2 with high transfection efficiency) significantly suppresses erastin-induced cytotoxicity/damages and partially abolishes the hepatoprotective effects of genipin in L-O2 cells (Fig. 7E).

Genipin exerts hepatoprotective effects by inhibiting ALOX15-launched lipid peroxidation. (A, B) The KEGG pathway analysis using liver tissues of mice subjected to CCl4 intoxication with and without genipin in terms of RNA-sequencing. (C) Evaluation of several oxylipins associated with ALOX15 catabolism including AA, 15-HpETE and 15-HETE in terms of UHPLC-MS/MS. (D) Determination of protein levels of ALOX15 in L-O2 cells transfected with NC siRNA or ALOX15 siRNA. (E) Assessment of the viability of L-O2 cells and the accumulation of LDH in L-O2 cells transfected with NC siRNA or ALOX15 siRNA. ###p < 0.001 vs. control group, &&&p < 0.001 vs. erastin group, *** p < 0.001 vs. ALOX15 siRNA group
Discussion
Our findings in the current study implicated that genipin, a natural ingredient extracted from Gardenia jasminoides Ellis, exhibited hepatoprotective effects by abrogating ferroptosis in the context of CCl4-triggerd acute liver injury. Moreover, mechanistic research revealed that a cascade of ferroptosis-associated events is influenced upon genipin administration in the injurious mice livers. Also, in-depth investigation regarding molecular mechanisms indicated that pharmacological modification of dysregulated GPX4 and ALOX15-launched lipid peroxidation accounts for medicinal effects of genipin to be a promising therapeutic approach for specific liver pathology. Previously, we denoted that genipin counteracts hepatotoxicity by enhancing autophagy among toxicant injury models [18]. In alignment with lately published report, genipin may exhibit its protective effects by modifying divergent modalities in relation to regulated cell death. It is tempting to target ferroptotic cell death in conjunction with impaired autophagic flux to potentiate hepatic damages recovery.
In the current study, we elaborate on the role of GPX4 dysregulation in the context of toxicant-induced ferroptosis. Previously, we argued that GPX4 and xCT serve as essential components to counteract the generation of specific lipid hydroperoxides in the presence of overwhelming iron load [13]. Inactivation of GPX4 or depletion of cellular GSH, a cofactor indispensible for selenocysteine-containing enzyme, can instigate a cascade of ferroptosis-associated events. Although acetaminophen (APAP)-triggered liver injury has been under extensive investigations for decades, the relationship between ferroptosis and APAP hepatotoxicity is still under debate. Particularly, Jaeschke H et al. argued that there is a lack of credible evidence to support the notion that APAP elicits ferroptotic hepatocellular death, taking into consideration that superoxide dismutase mimetics exhibit highly effective protection against APAP hepatotoxicity [30]. The CCl4-triggerd acute liver injury represents typical hallmarks in relation to ferroptosis, which is characterized by intensively oxidative stress, lipid peroxidation and expression of Ptgs2/p53 in addition to GSH consumption [31, 32]. As for oxidative stress phase, CCl4-derived toxicants and deleterious metabolites give rise to disruption and impairment to antioxidative defense systems including GPX members and GSH [33, 34]. For instance, Shah et al. found that over-production of ROS in CCl4-induced hepatic dysfunction model may result in protein inactivation manifested as a significant decrease pertaining to the levels of GPX [34]. Another report implicated that toxic metabolites of CCl4 (e.g., ●CCl3) can considerably impact the activity of indicative glutathione-metabolizing enzyme, meanwhile, direct reaction with sulfhydryl groups of GSH was responsible for its reduced functionality and concentration [33]. Accordingly, our findings aligned with aforesaid data in terms of significant reduction of GPX4 protein levels, which was restored upon genipin supplementation (Fig. 4A). Moreover, it was highlighted that there is a similar pattern concerning pertubation in xCT protein levels. Mounting evidence indicates that xCT-GSH-GPX4 signalings orchestrate tightly to exert antioxidant effects [35]. Notably, the negative regulation of xCT expression can be mainly attributed to transcription factor p53, mirrored as p53 diminishment contributing to the upregulation of xCT levels [26]. In the present investigation, we observed a remarkable increase of p53 protein level due to CCl4 intoxication, while genipin effectively restored its expression. Therefore, the synergistic effect of upregulated xCT-GPX4 pathway partially accounts for the hepatoprotective activities of genipin. As for lipid peroxidation phase, unneutralized CCl4 radicals orchestrating a covalent link with membranous proteins/lipids and mitochondria in hepatocytes to produce plenty of lipid radicals, all of which lead to the morphological and functional pertubations in liver cells [4, 36]. Given that GPX4 serves as the core inhibitor of ferroptosis, capable of eliminating lipid peroxides, and in consequence determines cell fate, the molecular docking analysis clearly supported a potential molecule interactions between genipin and GPX4 protein. Intriguingly, recent study reported exactly similar findings to the present investigation: apigenin supplementation, a natural plant flavonoid, repressed the levels of ROS and concentrations of MDA alongside recovery manifesting a combined decrease of GPX4 and xCT in di(2-ethylhexyl) phthalate-induced liver injury in the context of ferroptosis execution [37]. In that study, further molecular docking also revealed an interaction between the ligand apigenin and the amino acid residues of GPX4. On the other hand, Li et al. identified a series of substituted compound on the basis of combined computational (i.e., molecular docking program) and experimental screen, one of which activates GPX4 to serve as antioxidative (inhibition of intracellular ROS), anti-inflammatory (repression of AA oxidation) and cytoprotective (suppression of ferroptosis) agent [38]. Consistently, our results denoted that genipin supplementation effectively regulates a cascade of ferroptosis-associated events. The iron homeostasis, destruction of mitochondrial structure, production of lipid ROS and cell death were modulated upon genipin treatment in CCl4-intoxication models in light of determining catalytically active Fe2+, TEM imaging, BODIPY probe as well as TUNEL staining. These findings were also confirmed in vitro concerning multistage ferroptosis alongside GPX4 silencing experiments. Taken together, the hepatoprotective effects of genipin, to some extent, can be attributed to restore GPX4 expression levels aimed at preventing toxicant-induced ferroptosis.
It is well documented that lipid peroxidation represents hallmarks in liver pathology subjected to CCl4 challenge [39]. Plenty of bioactive carbonyls, such as MDA and 4-HNE, are produced in response to reaction between lipid peroxides and iron, all of which in turn conjugate with GSH and diminish intracellular GSH contents. Accordingly, augmented production of MDA and 4-HNE (both lipid peroxidation biomarkers) were observed in the livers of CCl4-triggered mice, which were remarkably attenuated by genipin administration. Furthermore, mechanistic research in terms of RNA-sequencing analysis unveiled that several gene clusters concerning biogenesis of unsaturated fatty acids, arachidonic acid metabolism and fatty acid elongation are substantially influenced by genipin treatment, pinpointing the key role of PUFAs metabolism in the context of ferroptotic cell death.
Lipoxygenase accounts for catalyzing the PUFAs peroxidation to corresponding hydroperoxy derivatives, resulting in the bursting of pro-ferroptotic oxidation process [40, 41]. ALOX15 is capable of using preferred substrates such as linoleic acid, docosahexaenoic acid and AA, which are incorporated into cholesterol esters/phospholipids or in free forms [42]. Of note, an n-6 PUFA, designated as AA, exhibits a major component of the membranous phospholipids, which is converted into 15-HpETE and 15-HETE by ALOX15 and metabolized towards various bioactive substances [43]. In agreement with these concepts, our results showed that genipin supplementation significantly downregulates ALOX15 protein expression in the CCl4-intoxicated livers. In this regard, UHPLC-MS/MS analysis denoted that AA, 15-HpETE and 15-HETE increase in toxicant injury models, and these shifts are reversed by genipin treatment. The participation of ALOX15 in erastin-induced ferroptosis was also validated by in vitro experiments. Taking into account the mechanism of lipid peroxidation as a controversy, these findings provide a novel therapeutic approach to target ALOX15-launched lipid peroxidation in divergent pathological conditions.
As for inflammatory response phase, CCl4-created free radicals are responsible for the hypertrophy and hyperplasia of macrophages in the livers, which can lead to damages to the hepatic parenchyma via massive release of detrimental and proinflammatory molecules [5, 44]. The results of the current study verified that intoxication of CCl4 is closely linked to aggravated inflammation. The macrophages identified by F4/80 staining and production of several cytokines (IL-1β, IL-6 and TNF-α) determined by Milliplex increased in CCl4 intoxication mice livers, whereas these alterations were reversed in the CCl4 + genipin group, coincided with findings in similar investigations by us and others [4, 18, 45]. As a matter of fact, the relationship between ALOX15 and inflammation appears to be intricate and heterogeneous. It is suggested that ALOX15 may considerably impact inflammatory response by producing lipid mediators [42]. Particularly, a recent report indicated that ALOX15-launched peroxidation of PUFA-phospholipids represents a pivotal initial event among ischemia injuries [29]. Taken together, in-depth investigations are warranted to elucidate underpinning molecular mechanisms regarding protective effects of genipin concerning interaction between lipid peroxidation and inflammation in the context of ferroptosis.
Genipin may influence a wide range of pathophysiological conditions/events to exert its hepatoprotective effects such as modification of mitochondria quality, anti-inflammatory activity, antioxidative activity, antifibrogenic effect, cholagogic effect and regulation of cell death [46]. On the other hand, accumulating evidence implicates that divergent forms of regulated cell death (e.g., apoptosis, necroptosis, autophagy and ferroptosis) can converge in pathogenic environment [47]. These modalities of regulated cell death mirror as the “backup” dying strategies to maintain internal homeostasis in the circumstance that the cellular death-triggering threshold is reached, since they may share overlapping mechanisms. The interplay between different forms of regulated cell death appears to be complicated and still under debate. For instance, autophagy can facilitate ferroptosis due to iron overloaded suppression of xCT, and erastin-induced ferroptosis was inhibited by removing Atg5/Atg7 [48, 49]. Conversely, a latest report showed that astaxanthin can protect against APAP-induced liver injury through inhibiting ferroptosis and promoting autophagy via the Nrf2/HO-1 pathway [50]. In conjunction with other reports, wherein the medicinal effects pertinent to genipin encompassed the induction of autophagy and activation of GPX4-Nrf2 axis, as key defensive systems against liver injury in the context of ferroptosis, we suppose it is operational to investigate the reciprocal relations between divergent forms of regulated cell death in the future studies [51].
Conclusion
In conclusion, our findings demonstrated that genipin treatment protects against CCl4-triggered acute liver injury by abrogating ferroptosis-dictated hepatocyte death, and pharmacological modification of dysregulated GPX4 and ALOX15-launched lipid peroxidation partially accounted for its medicinal effects as underlying molecular basis.
Data Availability
The datasets used or analyzed during the current study are available from the corresponding author on reasonable request.
References
Tak J, Kim YS, Kim TH, Park GC, Hwang S, Kim SG (2022) Galpha12 overexpression in hepatocytes by ER stress exacerbates acute liver injury via ROCK1-mediated miR-15a and ALOX12 dysregulation. Theranostics 12:1570–1588
Zhao T, Mao L, Yu Z, Hui Y, Feng H, Wang X, Lin L, Fan X, Chen X, Wang B, Cao X, Sun C (2021) Therapeutic potential of bicyclol in liver diseases: Lessons from a synthetic drug based on herbal derivative in traditional chinese medicine. Int Immunopharmacol 91:107308
Cheng N, Ren N, Gao H, Lei X, Zheng J, Cao W (2013) Antioxidant and hepatoprotective effects of Schisandra chinensis pollen extract on CCl4-induced acute liver damage in mice. Food Chem Toxicol 55:234–240
Popovic D, Kocic G, Katic V, Zarubica A, Velickovic LJ, Nickovic VP, Jovic A, Veljkovic A, Petrovic V, Rakic V, Jovic Z, Ulrih NP, Sokolovic D, Stojanovic M, Stankovic M, Radenkovic G, Nikolic GR, Lukac capital A C, Milosavljevic A, Sokolovic D (2019) Anthocyanins Protect Hepatocytes against CCl4-Induced Acute Liver Injury in Rats by Inhibiting Pro-inflammatory mediators, Polyamine Catabolism, Lipocalin-2, and Excessive Proliferation of Kupffer Cells. Antioxidants (Basel) 8
Ahn M, Kim J, Bang H, Moon J, Kim GO, Shin T (2016) Hepatoprotective effects of allyl isothiocyanate against carbon tetrachloride-induced hepatotoxicity in rat. Chem Biol Interact 254:102–108
Unsal V, Cicek M, Sabancilar I (2021) Toxicity of carbon tetrachloride, free radicals and role of antioxidants. Rev Environ Health 36:279–295
Doll S, Freitas FP, Shah R, Aldrovandi M, da Silva MC, Ingold I, Goya Grocin A, Xavier da Silva TN, Panzilius E, Scheel CH, Mourao A, Buday K, Sato M, Wanninger J, Vignane T, Mohana V, Rehberg M, Flatley A, Schepers A, Kurz A, White D, Sauer M, Sattler M, Tate EW, Schmitz W, Schulze A, O’Donnell V, Proneth B, Popowicz GM, Pratt DA, Angeli JPF, Conrad M (2019) FSP1 is a glutathione-independent ferroptosis suppressor. Nature 575:693–698
Stockwell BR, Jiang X, Gu W (2020) Emerging mechanisms and Disease Relevance of Ferroptosis. Trends Cell Biol 30:478–490
Fujii J, Homma T, Osaki T (2022) Superoxide Radicals in the execution of cell death. Antioxid (Basel) 11
Kagan VE, Mao G, Qu F, Angeli JP, Doll S, Croix CS, Dar HH, Liu B, Tyurin VA, Ritov VB, Kapralov AA, Amoscato AA, Jiang J, Anthonymuthu T, Mohammadyani D, Yang Q, Proneth B, Klein-Seetharaman J, Watkins S, Bahar I, Greenberger J, Mallampalli RK, Stockwell BR, Tyurina YY, Conrad M, Bayir H (2017) Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat Chem Biol 13:81–90
Ajoolabady A, Tang D, Kroemer G, Ren J (2022) Ferroptosis in hepatocellular carcinoma: mechanisms and targeted therapy. Br J Cancer.
Ma C, Han L, Zhu Z, Heng Pang C, Pan G (2022) Mineral metabolism and ferroptosis in non-alcoholic fatty liver diseases. Biochem Pharmacol 205:115242
Mao L, Zhao T, Song Y, Lin L, Fan X, Cui B, Feng H, Wang X, Yu Q, Zhang J, Jiang K, Wang B, Sun C (2020) The emerging role of ferroptosis in non-cancer liver diseases: hype or increasing hope? Cell Death Dis 11:518
Qi J, Kim JW, Zhou Z, Lim CW, Kim B (2020) Ferroptosis affects the progression of nonalcoholic steatohepatitis via the modulation of lipid peroxidation-mediated cell death in mice. Am J Pathol 190:68–81
Dai C, Li H, Wang Y, Tang S, Velkov T, Shen J (2021) Inhibition of oxidative stress and ALOX12 and NF-kappaB pathways contribute to the Protective Effect of Baicalein on Carbon Tetrachloride-Induced Acute Liver Injury. Antioxid (Basel) 10
Tsai WH, Yang CC, Li PC, Chen WC, Chien CT (2013) Therapeutic potential of traditional chinese medicine on inflammatory diseases. J Tradit Complement Med 3:142–151
Yim JS, Kim YS, Moon SK, Cho KH, Bae HS, Kim JJ, Park EK, Kim DH (2004) Metabolic activities of ginsenoside Rb1, baicalin, glycyrrhizin and geniposide to their bioactive compounds by human intestinal microflora. Biol Pharm Bull 27:1580–1583
Wang Y, Zhao T, Deng Y, Hou L, Fan X, Lin L, Zhao W, Jiang K, Sun C (2019) Genipin Ameliorates Carbon Tetrachloride-Induced Liver Injury in Mice via the Concomitant Inhibition of Inflammation and Induction of Autophagy. Oxid Med Cell Longev 2019:3729051
Chen X, Kang R, Kroemer G, Tang D (2021) Broadening horizons: the role of ferroptosis in cancer. Nat Rev Clin Oncol 18:280–296
Cho HI, Kim SJ, Choi JW, Lee SM (2016) Genipin alleviates sepsis-induced liver injury by restoring autophagy. Br J Pharmacol 173:980–991
Yamada N, Karasawa T, Wakiya T, Sadatomo A, Ito H, Kamata R, Watanabe S, Komada T, Kimura H, Sanada Y, Sakuma Y, Mizuta K, Ohno N, Sata N, Takahashi M (2020) Iron overload as a risk factor for hepatic ischemia-reperfusion injury in liver transplantation: potential role of ferroptosis. Am J Transplant 20:1606–1618
Stancic A, Velickovic K, Markelic M, Grigorov I, Saksida T, Savic N, Vucetic M, Martinovic V, Ivanovic A, Otasevic V (2022) Involvement of ferroptosis in Diabetes-Induced Liver Pathology. Int J Mol Sci 23
Chen J, Li X, Ge C, Min J, Wang F (2022) The multifaceted role of ferroptosis in liver disease. Cell Death Differ 29:467–480
Tang D, Chen X, Kang R, Kroemer G (2021) Ferroptosis: molecular mechanisms and health implications. Cell Res 31:107–125
Liu T, Shu J, Liu Y, Xie J, Li T, Li H, Li L (2022) Atorvastatin attenuates ferroptosis-dependent myocardial injury and inflammation following coronary microembolization via the Hif1a/Ptgs2 pathway. Front Pharmacol 13:1057583
Jiang L, Kon N, Li T, Wang SJ, Su T, Hibshoosh H, Baer R, Gu W (2015) Ferroptosis as a p53-mediated activity during tumour suppression. Nature 520:57–62
Tarangelo A, Dixon S (2018) The p53-p21 pathway inhibits ferroptosis during metabolic stress. Oncotarget 9:24572–24573
Yang WS, Stockwell BR (2008) Synthetic lethal screening identifies compounds activating iron-dependent, nonapoptotic cell death in oncogenic-RAS-harboring cancer cells. Chem Biol 15:234–245
Ma XH, Liu JH, Liu CY, Sun WY, Duan WJ, Wang G, Kurihara H, He RR, Li YF, Chen Y, Shang H (2022) ALOX15-launched PUFA-phospholipids peroxidation increases the susceptibility of ferroptosis in ischemia-induced myocardial damage. Signal Transduct Target Ther 7:288
Jaeschke H, Adelusi OB, Ramachandran A (2021) Ferroptosis and Acetaminophen Hepatotoxicity: are we going down another rabbit. Hole? Gene Expr 20:169–178
Huang GJ, Deng JS, Chiu CS, Liao JC, Hsieh WT, Sheu MJ, Wu CH (2012) Hispolon Protects against Acute Liver Damage in the Rat by Inhibiting Lipid Peroxidation, Proinflammatory Cytokine, and Oxidative Stress and Downregulating the Expressions of iNOS, COX-2, and MMP-9. Evid Based Complement Alternat Med 2012:480714
Zahedi K, Barone SL, Xu J, Steinbergs N, Schuster R, Lentsch AB, Amlal H, Wang J, Casero RA Jr, Soleimani M (2012) Hepatocyte-specific ablation of spermine/spermidine-N1-acetyltransferase gene reduces the severity of CCl4-induced acute liver injury. Am J Physiol Gastrointest Liver Physiol 303:G546–560
Popovic D, Kocic G, Katic V, Jovic Z, Zarubica A, Jankovic Velickovic L, Nikolic V, Jovic A, Kundalic B, Rakic V, Ulrih NP, Skrt M, Sokolovic D, Dinic L, Stojanovic M, Milosavljevic A, Velickovic F, Sokolovic D (2019) Protective effects of anthocyanins from bilberry extract in rats exposed to nephrotoxic effects of carbon tetrachloride. Chem Biol Interact 304:61–72
Shah MD, Gnanaraj C, Haque AT, Iqbal M (2015) Antioxidative and chemopreventive effects of Nephrolepis biserrata against carbon tetrachloride (CCl4)-induced oxidative stress and hepatic dysfunction in rats. Pharm Biol 53:31–39
Yuan Y, Yucai L, Lu L, Hui L, Yong P, Haiyang Y (2022) Acrylamide induces ferroptosis in HSC-T6 cells by causing antioxidant imbalance of the XCT-GSH-GPX4 signaling and mitochondrial dysfunction. Toxicol Lett 368:24–32
Knockaert L, Berson A, Ribault C, Prost PE, Fautrel A, Pajaud J, Lepage S, Lucas-Clerc C, Begue JM, Fromenty B, Robin MA (2012) Carbon tetrachloride-mediated lipid peroxidation induces early mitochondrial alterations in mouse liver. Lab Invest 92:396–410
Han D, Yao Y, Chen L, Miao Z, Xu S (2022) Apigenin ameliorates di(2-ethylhexyl) phthalate-induced ferroptosis: the activation of glutathione peroxidase 4 and suppression of iron intake. Food Chem Toxicol 164:113089
Li C, Deng X, Xie X, Liu Y, Friedmann Angeli JP, Lai L (2018) Activation of glutathione peroxidase 4 as a novel anti-inflammatory strategy. Front Pharmacol 9:1120
Wu SJ, Lin YH, Chu CC, Tsai YH, Chao JC (2008) Curcumin or saikosaponin a improves hepatic antioxidant capacity and protects against CCl4-induced liver injury in rats. J Med Food 11:224–229
Kuhn H, Humeniuk L, Kozlov N, Roigas S, Adel S, Heydeck D (2018) The evolutionary hypothesis of reaction specificity of mammalian ALOX15 orthologs. Prog Lipid Res 72:55–74
Yang WS, Kim KJ, Gaschler MM, Patel M, Shchepinov MS, Stockwell BR (2016) Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc Natl Acad Sci U S A 113:E4966–4975
Singh NK, Rao GN (2019) Emerging role of 12/15-Lipoxygenase (ALOX15) in human pathologies. Prog Lipid Res 73:28–45
Markworth JF, Mitchell CJ, D’Souza RF, Aasen KMM, Durainayagam BR, Mitchell SM, Chan AHC, Sinclair AJ, Garg M, Cameron-Smith D (2018) Arachidonic acid supplementation modulates blood and skeletal muscle lipid profile with no effect on basal inflammation in resistance exercise trained men. Prostaglandins Leukot Essent Fatty Acids 128:74–86
Torres LR, Santana FC, Torres-Leal FL, Melo IL, Yoshime LT, Matos-Neto EM, Seelaender MC, Araujo CM, Cogliati B, Mancini-Filho J (2016) Pequi (Caryocar brasiliense Camb.) Almond oil attenuates carbon tetrachloride-induced acute hepatic injury in rats: antioxidant and anti-inflammatory effects. Food Chem Toxicol 97:205–216
Chan CC, Lee KC, Huang YH, Chou CK, Lin HC, Lee FY (2014) Regulation by resveratrol of the cellular factors mediating liver damage and regeneration after acute toxic liver injury. J Gastroenterol Hepatol 29:603–613
Fan X, Lin L, Cui B, Zhao T, Mao L, Song Y, Wang X, Feng H, Qingxiang Y, Zhang J, Jiang K, Cao X, Wang B, Sun C (2020) Therapeutic potential of genipin in various acute liver injury, fulminant hepatitis, NAFLD and other non-cancer liver diseases: more friend than foe. Pharmacol Res 159:104945
You H, Wang L, Bu F, Meng H, Huang C, Fang G, Li J (2022) Ferroptosis: Shedding Light on Mechanisms and Therapeutic Opportunities in Liver Diseases. Cells 11
Hou W, Xie Y, Song X, Sun X, Lotze MT, Zeh HJ 3rd, Kang R, Tang D (2016) Autophagy promotes ferroptosis by degradation of ferritin. Autophagy 12:1425–1428
Song X, Zhu S, Chen P, Hou W, Wen Q, Liu J, Xie Y, Liu J, Klionsky DJ, Kroemer G, Lotze MT, Zeh HJ, Kang R, Tang D (2018) AMPK-Mediated BECN1 phosphorylation promotes ferroptosis by directly blocking System Xc(-) activity. Curr Biol 28:2388–2399e2385
Cai X, Hua S, Deng J, Du Z, Zhang D, Liu Z, Khan NU, Zhou M, Chen Z (2022) Astaxanthin activated the Nrf2/HO-1 pathway to Enhance Autophagy and inhibit ferroptosis, ameliorating Acetaminophen-Induced Liver Injury. ACS Appl Mater Interfaces 14:42887–42903
Liu J, Huang C, Liu J, Meng C, Gu Q, Du X, Yan M, Yu Y, Liu F, Xia C (2022) Nrf2 and its dependent autophagy activation cooperatively counteract ferroptosis to alleviate acute liver injury. Pharmacol Res 187:106563
Funding
The authors declare that no funds, grants, or other support were received during the preparation of this manuscript.
Author information
Authors and Affiliations
Contributions
Conception or design of the work: Xiaofei Fan, Xiaoyu Wang, Yangyang Hui and Chao Sun; Data collection: Tianming Zhao, Lihong Mao, Binxin Cui and Weilong Zhong; Drafting the article and critical revision of the article: Chao Sun; Final ap-proval of the version to be published: all authors.
Corresponding author
Ethics declarations
Ethical approval
All animal studies were approved by the Institutional Animal Care and Use Committee at Tianjin Medical University General Hospital (IRB2021-DWFL-142).
Competing interest
All authors declare no conflict of interest.
Additional information
Xiaofei Fan, Xiaoyu Wang and Yangyang Hui contributed equally to this work.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Fan, X., Wang, X., Hui, Y. et al. Genipin protects against acute liver injury by abrogating ferroptosis via modification of GPX4 and ALOX15-launched lipid peroxidation in mice. Apoptosis 28, 1469–1483 (2023). https://doi.org/10.1007/s10495-023-01867-9
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10495-023-01867-9