
Abstract
Although radiation-induced bystander effects have been broadly explored in various biological systems, the molecular mechanisms and the consequences of different regulatory factors (dose, time, cell type) on bystander responses are not clearly understood. This study investigates the effects of irradiated cell-conditioned media (ICCM) collected at different times post-irradiation on bystander cancer cells regarding DNA damage and apoptosis induction. Human hepatocellular carcinoma HepG2 cells were exposed to γ-ray doses of 2 Gy, 5 Gy, and 8 Gy. In the early and late stages (1 h, 2 h, and 24 h) after irradiation, the ICCM was collected and transferred to unirradiated cells. Compared to control, bystander cells showed an increased level of H2AX phosphorylation, mitochondrial membrane depolarization, and elevation of intrinsic apoptotic pathway mediators such as p53, Bax, cas9, cas-3, and PARP cleavage. These results were confirmed by phosphatidylserine (PS) externalization and scanning electron microscopic observations, suggesting a rise in bystander HepG2 cell apoptosis. Anti-apoptotic Bcl2-level and viability were lower in bystander cells compared to control. The highest effects were observed in 8 Gy γ radiation-induced bystander cells. Even though the bystander effect was persistent at all time points of the study, ICCM at the early time points (1 or 2 h) had the most significant impact on the apoptosis markers in bystander cells. Nevertheless, 24 h ICCM induced the highest increase in H2AX and p53 phosphorylation and Bax levels. The effects of ICCM of irradiated HepG2 cells were additionally studied in normal liver cells BRL-3A to simulate actual radiotherapy conditions. The outcomes suggest that the expression of the signaling mediators in bystander cells is highly dynamic. A cross-talk between those signaling mediators regulates bystander responses depending on the radiation dose and time of incubation post-irradiation.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The interaction between ionizing radiation (IR) and cells induces the development of repair-resistant damage clusters. These irreparable DNA damages cause mutations, genomic instability, inflammation, oxidative stress, and activate cell death processes, including apoptosis, necrosis, and senescence [1]. Recent reports suggest that these processes promote the release of danger signals or Damage-Associated Molecular Patterns (DAMPs) from irradiated cells, like, cytokines, interleukins, chemokines, ROS, DNA/RNA fragments, or other small proteins [2, 3]. These stress signals activate secondary responses and oxidative stress-induced complications in radiation non-targeted cells. Transmission of the signals between irradiated and unirradiated cells occurs via either gap junctions or soluble signaling mediators. The resultant damages in adjacent non-targeted cells, broadly termed radiation-induced bystander effects (RIBE), include death, chromosomal damages, mutations, with modifications in gene expression and protein levels [4, 5]. In recent years, RIBE has gained importance, not only because of its clinical significance during radiotherapy but additionally due to the diversity of findings depending on various biological (cell types) and physical factors (such as time, radiation dose, LET) [4].
Cellular death is a crucial endpoint that has been explored in cells exposed to radiation. The highly regulated type of cell death, apoptosis, is widely triggered by radiation in cancer cells, especially the intrinsic pathway of apoptosis [6]. Radiation has an immense impact on cellular morphology and induces considerable modifications in the status of apoptosis markers. Exposure to radiation causes irreparable DNA damages, especially double-stranded breaks, leading to the accumulation of p53 protein [7]. p53 stimulates the expression of pro-apoptotic mediators, thereby disrupting the intricate balance between cellular anti-apoptotic and pro-apoptotic proteins. p53 dissolves the complex of anti-apoptotic Bcl2 and the pro-apoptotic Bax. Released Bax activates cell death by stimulating the release of cytochrome c (cyt c) from mitochondria [8, 9]. Radiation reduces mitochondrial membrane potential (∆ψm) [10], which is one of the factors that stimulate Bax translocation to the mitochondria [11]. Bax forms transmembrane pores and activates the mitochondrial permeability transition pores (MPTP) [12]. MPTP opening boosts the passage of ions and molecules, resulting in the decoupling of the respiratory chain and cyt c release into the cytosol. Cyt c, APAF1 (apoptotic protease activating factor 1) and initiator caspase-9 (cas-9) form apoptosome complex. The initiator cas-9 triggers the effector caspases, like caspase-3 (cas-3), inducing cell death [8, 9]. Effector caspases cleave substrates involved with apoptosis regulation. Poly (ADP-ribose) polymerase (PARP) is a DNA damage repair protein that activates DNA repair pathways in response to radiation exposure. PARP is a substrate of cas-3, and proteolytic cleavage of PARP is usually considered a hallmark of apoptosis [13, 14].
Even though the mechanism of IR-induced cell death in irradiated cells is known, there is a dearth of information regarding the mechanism of bystander cell death under the influence of radiation. The study of IR-induced apoptosis and the underlying molecular mediators in bystander cells are crucial in radiation biology to apprehend the severity of damages caused by IR in non-targeted cells. It is crucial to understand whether similar signaling pathways are activated, and the status of apoptosis markers is altered similarly between irradiated and bystander cells. The bystander response is often more complex than the direct response. The existing literature highlights the intrinsic pathway of apoptosis as the predominant mode of bystander cell death [2, 15,16,17,18,19]. Experimentally bystander effects are studied either by media transfer technique or co-culture with irradiated cells. The irradiated cell-conditioned media (ICCM) is transferred to the bystander cells in the media transfer technique. In vitro studies have highlighted that in the same cell type, ICCM transfer induces different effects under different radiation doses [20, 21]. These conflicting results hint at the complex mechanisms regulating bystander effects, yet to be determined. Different cell types respond differently to bystander signaling [22, 23]. However, the primary question that remains to be answered is how the different signaling mediators in bystander cells interact and cross-talk with one other to generate bystander responses like apoptosis at different radiation doses and at early and late time points post-irradiation.
RIBE is an important concept in the field of radiation biology. Focusing on how and why bystander cells die can provide new insights into RIBE. This study aims to unravel the effects of ionizing gamma (γ) rays at different doses (2 Gy, 5 Gy, and 8 Gy) on bystander radio-resistant hepatocellular carcinoma cells (HepG2), with emphasis on DNA damage and apoptosis induction, morphological alterations, and the status of the signaling mediators of the intrinsic apoptotic pathway. The effects in bystander HepG2 cells have been analyzed compared to bystander normal liver cells (BRL-3A) to comprehensively evaluate the impact of radiation on bystander cell apoptosis. The work is designed to focus on the effect of ICCM collected at early (1 h, 2 h) and delayed time points (24 h) to obtain a more explicit expression profile of apoptosis markers.
Materials and methods
Cell culture
The human liver cancer cell line (HepG2) and normal buffalo rat liver cell line (BRL3A) were obtained from the National Centre for Cell Science (NCCS) in Pune, India. The cells were maintained in Dulbecco’s modified Eagle medium (DMEM) (Gibco, USA) containing 10% Fetal bovine serum (FBS) (Gibco, USA) and supplemented with Anti-Anti (100 ×) Antibiotic–Antimycotic (Gibco, USA) under sterile conditions (Class II Biological Safety Cabinet, Esco, Singapore) and grown at 37 °C in a humidified incubator (NuAire, USA) with 95% air and 5% CO2.
Cell irradiation and transfer of conditioned medium
The cells were seeded for different experiments in 60 mm or 100 mm Petri dishes (HiMedia, USA) at a seeding density of 0.8 × 106 and 2.2 × 106, respectively, using the Olympus Automated Cell Counter model R1 (Olympus, Japan). After adherent (24 h), HepG2 cells were irradiated at γ ray doses of 2 Gy, 5 Gy, and 8 Gy at a dose rate of 1.27 kGy/h using a Co60 PLC-based Gamma (GC 1200) irradiation system (UGC-DAE Consortium for Scientific Research, Kolkata Centre, India). After 1, 2, or 24 h of irradiation, the conditioned media (ICCM) from irradiated HepG2 cells were collected and filtered through 0.22 μm filters (Millipore, USA). In an incubator (NuAire, USA), the non-irradiated HepG2 and BRL-3A cells were cultured with the filtered, conditioned media for 1 h at 37 °C and 5% CO2. Post incubation, the bystander cells were ready for experiments.
As reported earlier by our group [4], bystander signaling is different between two cancer cells and between cancer and normal cells, with cancer cells showing more severe effects. Proliferative cancer cells largely remain in the S-phase of the cell cycle and possess mutated DNA damage repair systems, making them more vulnerable to bystander genotoxic effects. The effects of irradiated normal cells on bystander cells are less noticeable because normal cells possess pronounced repair systems. During radiotherapy, stress signals released from radiation-exposed cancer cells affect vicinal cancer and normal cells, considered bystanders. The release of stress signals is highly prominent in irradiated cancer cells because this indicates cancer cell survival strategy to promote growth, progression, and angiogenesis. To better understand the communication between irradiated cancer cells and bystander cancer and normal cells, only the ICCM of irradiated HepG2 cells was chosen as the stimulus condition in the present study.
Experimental design
The cells were divided into the following experimental groups. Experiments were carried out on each group to determine the effects of different doses of γ-rays at different time points post-irradiation (Fig. 1).

Schematic representation of the experimental design for this study. The ICCM from irradiated HepG2 cells at each dose (2, 5, 8 Gy) was transferred to bystander HepG2 and BRL-3A cells at three different time points (1, 2, 24 h) post-irradiation. The irradiated cells at each time point served as the radiation control of the respective bystander cells. There were 18 bystander experiment groups, nine corresponding irradiation controls. There were non-irradiated controls for every time point. The figure was created using images from Servier Medical Art Commons Attribution 3.0 Unported License. (http://smart.servier.com). Servier Medical Art by Servier is licensed under a Creative Commons Attribution 3.0 Unported License
-
(i)
Control group: HepG2 cells without any treatment.
-
(ii)
Irradiation groups
-
A.
2 Gy irradiation (2 Gy) group: HepG2 cells treated with 2 Gy dose of γ-ray. The ICCM were collected at 1 h, 2 h, and 24 h post-irradiation
-
B.
5 Gy irradiation (5 Gy) group: HepG2 cells treated with 5 Gy dose of γ-ray. The ICCM were collected at 1 h, 2 h, and 24 h post-irradiation
-
C.
8 Gy irradiation (8 Gy) group: HepG2 cells treated with 8 Gy dose of γ-ray. The ICCM were collected at 1 h, 2 h, and 24 h post-irradiation.
-
A.
-
(iii)
Bystander groups
-
A.
2 Gy bystander (2 By) group: Non-irradiated HepG2 and BRL-3A cells receiving 1 h, 2 h, and 24 h ICCM from 2 Gy group
-
B.
5 Gy bystander (5 By) group: Non-irradiated HepG2 and BRL-3A cells receiving 1 h, 2 h, and 24 h ICCM from 5 Gy group
-
C.
8 Gy bystander (8 By) group: Non-irradiated HepG2 and BRL-3A cells receiving 1 h, 2 h, and 24 h ICCM from 8 Gy group.
-
A.
Trypan blue exclusion assay and MTT assay showed the LD50 dose of γ-ray on HepG2 cells as 8 Gy. Since the effect of irradiated cell-conditioned media (ICCM) was studied on the bystander cells, the LD50 was determined only in the case of γ-irradiated HepG2 cells. Analysis of HepG2 cell viability at different radiation doses, determination of the LD50 dose, and careful review of research work [23,24,25,26,27,28,29,30] on bystander effects corroborated the selection of 2 Gy, 5 Gy, and 8 Gy doses of γ-radiation for the study.
Cell viability assay
HepG2 cell viability was estimated using trypan blue exclusion assay and MTT assay. Dead cells with distorted cell membranes take up trypan blue dye and are determined visually by their blue cytoplasm. Living cells exclude the dye and possess clear cytoplasm. 100 μl of trypan blue stock solution (0.4%) was mixed with 100 μl of cells. Cells were loaded in a hemocytometer and counted using Olympus Automated Cell Counter model R1 (Olympus, Japan). % viable cells was calculated as [1 − (Number of dead cells/total number of cells)] × 100. Metabolically active cells can reduce MTT to insoluble, dark purple-colored formazan products. A 5 mg/ml MTT [3-(4,5-Dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide] (Merck, USA) stock solution was prepared using (1 ×) PBS. Cells were dispersed within 96-well microtiter plates (HiMedia, USA) (~ 30,000 cells/well). A 24-h pre-incubation time was allowed before irradiation. Changing the culture medium from the wells with MTT solution (10 µl), the cells were incubated at 37 °C and 5% CO2 for 3 h. Cells were inspected to confirm the reduction of the tetrazolium. MTT solution was then removed, and 100 µl of DMSO (Merck, USA) was added to each well to dissolve the formazan crystals. Plates were shaken for 15 min to ensure good solubilization. Absorbance readings on each well were measured at 540 nm on UV/Vis Spectrophotometer (Hitachi, Japan). The percentage cell viability was determined using the formula: (OD of sample/OD of control) × 100%.
Determination of alterations in mitochondrial membrane potential
Alterations in mitochondrial membrane potential (ΔΨm) in HepG2 cells were studied using JC1 (5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolylcarbocyanine iodide) dye. Mitochondrial disruption is an important feature of the early stages of apoptosis. In cells with normal ΔΨm, JC-1 dye accumulates in mitochondria, forming red fluorescent JC1 aggregates (~ 590 nm). Due to increased mitochondrial membrane permeability and loss of electrochemical potential in apoptotic cells, JC1 retains original green fluorescence (~ 529 nm). Mitochondrial depolarization is thus indicated by a decrease in the red/green fluorescence intensity ratio. Post-treatment and incubation, cells were washed with (1X) PBS twice and trypsinized (0.25% Trypsin–EDTA, Gibco, USA). The detached cells and media were subjected to centrifugation (Sorvall RC6 Plus Centrifuge, Thermo Scientific, USA) at 3000 rpm for 3 min, and the supernatant in each tube (1.5 ml micro-centrifuge tube, Tarson, India) were discarded. 200 μM JC-1 stock solution (MitoProbe™ JC-1 Assay Kit, Invitrogen, Thermo Fisher Scientific, USA) was prepared freshly. 10 µl of JC1 stock solution was added to each tube and incubated at 37 °C, 5% CO2 for 30 min. Cells were washed with 1(×) PBS and re-suspended in 500 µl of JC-1 binding buffer (1 ×). These samples were analyzed by flow cytometry (BD FACS Calibur, Becton, Dickinson, and Company, USA).
Detection of phosphatidylserine (PS) externalization using PI/annexin double staining
Translocation of phosphatidylserine (PS) from the inner to the outer surface of the plasma membrane is a marker for apoptosis. FLUOS-conjugated annexin-V and propidium iodide (PI) were used to detect and quantify apoptosis and differentiation from necrosis at the single-cell level (Annexin-V-FLUOS Staining Kit, Roche, Merck, Kenilworth, USA). Post-treatment and incubation, HepG2 cells were washed with (1 ×) PBS twice and trypsinized (0.25% Trypsin–EDTA, Gibco, USA). The detached cells were subjected to centrifugation (Sorvall RC6 Plus Centrifuge, Thermo Scientific, USA) at 3000 rpm for 3 min. The pellet in each tube was suspended with 100 µl incubation buffer and stained with 2 μl Annexin-V-FLUOS labeling reagent and 2 μl PI solution. Post incubation for 15 min, the cells were analyzed in the flow cytometer (BD FACS Calibur, Becton, Dickinson and Company, USA) to determine non-apoptotic (negative for both dyes), early apoptotic (Annexin + /PI−), late apoptotic (Annexin +/PI +), and necrotic (PI +) cells.
Flow cytometry analysis of protein expression
Following irradiation and incubation, the cells were trypsinized using 0.25% Trypsin–EDTA (Gibco, USA) and centrifuged (Sorvall RC6 Plus Centrifuge, Thermo Scientific, USA) at 3200×g for 5 min at 4 °C. The pelleted cells were fixed with 4% paraformaldehyde (Sigma Aldrich, USA) and permeabilized with 0.1% Triton X-100 (Sigma Aldrich, USA) for 5 min. Cells were washed twice with 1(×) PBS containing 3% FBS (Gibco, USA). The pellet was re-suspended in primary antibody (1:200 dilution), incubated on ice for 2 h, centrifuged at 2500 rpm at 4 °C for 5 min, and washed twice with 1 (×) PBS. Then cells were re-suspended in fluorescence tagged secondary antibodies (1:200 dilution) for 30 min on ice. The expressions of phospho histone H2AX [(Ser139) (20E3) Rabbit mAb, Cat# 9718, RRID:AB_2118009], phospho p53 [(Ser15) (16G8) Mouse mAb, Cat# 9286, RRID:AB_331741], Bax [(D2E11) Rabbit mAb, Cat# 5023, RRID:AB_10557411], phospho-Bcl-2 [(Ser70) (5H2) Rabbit mAb, Cat# 2827, RRID:AB_659950], cleaved Caspase-9 [(Asp330) (D2D4) Rabbit mAb, Cat# 7237, RRID:AB_10895832), cleaved Caspase-3 [(Asp175) (5A1E) Rabbit mAb, Cat# 9664, RRID:AB_2070042] and cleaved PARP [(Asp214) (D64E10) XP Rabbit mAb, Cat# 5625, RRID:AB_10699459] were analyzed following this protocol. Alexa Fluor tagged anti-rabbit and anti-mouse secondary antibodies [Anti-rabbit IgG (H + L), F(ab′)2 Fragment, Alexa Fluor® 488 Conjugate, Cat# 4412, RRID:AB_1904025], [Anti-mouse IgG (H + L), F(ab′)2 Fragment, Alexa Fluor® 488 Conjugate, Cat# 4408, RRID:AB_10694704] were used. All the antibodies were procured from Cell signaling technology, USA. Analysis was carried out in BD FACS Calibur machine equipped with Cell Quest Pro software.
Western blot analysis
50 µg of protein from different cell lysates (HepG2 and BRL-3A) were loaded in wells to analyze protein expression using western blot. The proteins were transferred to the activated PVDF membrane after separation using 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). The membrane with the transferred proteins was blocked with 5% bovine serum albumin (BSA) solution for 2 h and incubated with primary antibody (1:200 dilution) overnight at 4 °C. The expressions of phospho histone H2AX, phospho p53, Bax, phospho-Bcl-2, cleaved caspase-9, cleaved caspase-3, and cleaved PARP were analyzed. β-actin antibody was used as the loading control. Alkaline phosphatase tagged anti-rabbit and anti-mouse secondary antibodies (1:200 dilution) [Anti-rabbit IgG, AP-linked Antibody, Cat# 7054], [Anti-mouse IgG, AP-linked Antibody, Cat# 7056] were used. All the antibodies were procured from Cell signaling technology, USA. The protein bands were visualized using NBT-BCIP solution. The bands were analyzed using Syngene Gbox EF Gel Documentation System and GeneTools software with GeneSnap imaging for quantification.
Scanning electron microscopic (SEM) studies
HepG2 cells were seeded in surface-treated, sterile cell culture grade coverslips (HiMedia, USA). Post-treatment and incubation, cells were washed with PBS (1 ×) 3 times and fixed using 0.4% glutaraldehyde fixative (Sigma Aldrich, USA). Then the cells were kept at room temperature for 24 h. Post PBS (1 ×) wash, the cells were dehydrated through a 10% to 100% ethanol series. Next, 1:2 solution of hexamethyldisilazane (HMDS) (Sigma Aldrich, USA): 100% ethanol (Merck, USA) followed by 2:1 HMDS:ethanol and 100% HMDS were added in subsequent steps. The samples were immersed in absolute HMDS, covered loosely, and kept in a fume hood overnight. HMDS evaporated, leaving the sample ready for sputter coating and SEM imaging. SEM imaging (Zeiss Evo 18 special edition, Germany) was done at the Centre for Research in Nanoscience & Nanotechnology (CRNN), University of Calcutta, Kolkata.
Statistical analysis
All experiments were carried out in triplicate. The data are presented in terms of mean ± standard error (S.E.) of the mean. Statistical significance for group comparisons was determined using one-way analysis of variance (ANOVA) along with the post-hoc Tukey HSD test. Correlation between the variables was determined using the Pearson correlation coefficient formula. In all cases, a p-value < 0.05 was considered statistically significant. ‘*’ represented the significance of the difference between control vs. irradiated and control vs. bystander groups. ‘#’ represented the significance of difference in irradiated vs. bystander group and between bystander groups (*p < 0.05, **p < 0.01 / #p < 0.05, ##p < 0.01).
Results
γ-Radiation exposure decreased HepG2 cell survival, and the cell viability of bystander cells showed a negative relation with apoptotic and DNA damage markers
Trypan blue exclusion assay and MTT assay were used to select the radiation doses for the study. Data showed the LD50 dose of γ-radiation on HepG2 cells was around 8 Gy at 24 h post-irradiation showing 50.3 ± 1.82% viable cells (Fig. 2A, B). At 1 h or 2 h post-irradiation, 8 Gy cells showed 59.45 ± 2.16% and 58.93 ± 2.44% viable cells (Fig. 2A). Again, at 36 h or 48 h post 8 Gy γ-irradiation, percentage of viable cells increased to 62.78 ± 1.87% and 65.89 ± 2.23%, respectively (Fig. 2A). Since after 24 h of γ-irradiation, cell viability increased, 1 h, 2 h, and 24 h post-irradiation time points were chosen. The radiation-induced bystander effect being low dose phenomenon, 8 Gy, and lower doses of 2 Gy and 5 Gy were selected for the study (Fig. 2B).

The viability of irradiated and bystander HepG2 cells decreased under the influence of γ-rays. A HepG2 cells were irradiated with different γ-ray doses (up to 10 Gy) at different time points post-irradiation (up to 48 h), and cell viability was measured. Radiation doses were taken along the X-axis, and cell viability % was taken along the Y-axis. B MTT assay showed the LD50 dose of γ-radiation on HepG2 cells was 8 Gy. In the X-axis, radiation doses and in the Y-axis, percentage cell viability were plotted. C Percentage cell viability of the various experimental groups- bystander (2 By, 5 By, 8 By) and irradiated (2 Gy, 5 Gy, 8 Gy) groups decreased compared to control at different time points. D Cell viability of bystander cells at different doses and time points (1 h, 2 h, 24 h ICCM) in comparison to control. E–F Evaluation of the relationship between viability and cellular damage markers in (E). 1 h ICCM treated and F. 2 h ICCM treated 8 By cells. The bystander cell viability showed a significant, very small negative correlation with the different parameters. Values are mean ± S.E. ‘*’ represented significant difference compared to control. ‘#’ represented a significant difference in bystander effect compared to irradiated and bystander cells. (*p < 0.05, **p < 0.01, #p < 0.05, ##p < 0.01)
The MTT assay highlighted that under all treatment conditions, except in 1 h ICCM treated 2 By cells, the bystander cell viability was significantly reduced compared to the control (Fig. 2C, D). At a particular radiation dose, the percentage decrease in viability was independent of the time of ICCM transfer from irradiated cells (Fig. 2D). 8 By cells treated with 2 h ICCM showed the most substantial reduction in viability (Fig. 2D). However, except 2By cells, all other bystander cell groups demonstrated significantly higher viability than irradiated cells (Fig. 2C). In 1 h ICCM treated 8 By cells, a rise in viability was correlated with decreased PS externalization, cleaved cas-3, and cleaved PARP levels (Fig. 2E). While in 2 h ICCM treated 8 By cells, decrease in viability was linked to increased mitochondrial membrane depolarization, phospho-p53, phospho-H2AX, and Bax levels (Figs. 2F, 8B).
No significant association was noticed between reduced mitochondrial membrane potential (ΔΨm) and increased PS externalization in the bystander HepG2 cells
γ-rays induced considerable loss of mitochondrial membrane potential in bystander HepG2 cells compared to control (Fig. 3A–C). However, bystander cells except for 2 h ICCM treated 5 By cells demonstrated a significantly lower level of depolarization than irradiated cells (Fig. 3A–C). The most substantial depolarization was observed in 2 h ICCM treated 8 By cells, which was not significantly different from that in 5 By cells (Fig. 3D). In 8 By cells, post 1 h ICCM treatment, increase in mitochondrial membrane depolarization was correlated with a rise in Bax and cleaved cas-9 levels and decrease in phospho-Bcl2 levels, while in 2 h ICCM treated cells, a positive correlation was observed with phospho-p53, phospho-H2AX, Bax and cleaved cas-9 levels. However, in 24 h ICCM treated 8 By cells, a reduction in mitochondrial membrane potential was associated only with increased phospho-H2AX and Bax levels (Fig. 3E).

γ-rays induce mitochondrial membrane depolarization and phosphatidylserine externalization in bystander cells with the highest effects at the earlier time points. A–C Percentage of cells with red and green fluorescence at different doses and time points post-irradiation: A 1 h, B 2 h. C 24 h. The cells in the Q3 quadrant of the dot plots were viable cells with no loss of membrane potential. Apoptotic cells with mitochondrial membrane depolarization were present in the lower Q2 quadrant. The X-axis FL1-H channel (green) and the Y-axis FL2-H channel (Red) were taken. D The red to green fluorescence intensity ratio in bystander cells under different experimental conditions. A reduction in the red: green fluorescence ratio in the bar diagram indicated increased mitochondrial membrane depolarization. E Significant correlations between mitochondrial membrane depolarization and cellular damage markers in 1 h, 2 h, and 24 h ICCM treated 8 By cells. F–H HepG2 cells were stained with FLUOS-conjugated annexin-V and propidium iodide (PI). Cells in the Q2 quadrant were early apoptotic (Annexin + /PI−). Cells in the Q3 quadrant were late apoptotic (Annexin + /PI +), and the Q4 quadrant were necrotic (PI +) cells. The intensity of Annexin V in the FL4-H channel was taken along the X-axis, and the intensity of PI in the FL2-H channel was taken along Y-axis. PI-annexin study was carried out in control, irradiated, and bystander cells at different time points F 1 h, G 2 h, H 24 h post-irradiation. I The percentage of apoptosis (early and late) in bystander cells compared to control and irradiated cells at 1 h post-irradiation. J The time-dependent change in the percentage of bystander apoptotic cells at different radiation doses w.r.t. control. K Substantial positive correlations between PS externalization and cellular damage markers in 1 h, 2 h, and 24 h ICCM treated 8 By cells Values are mean ± S.E. ‘*’ represented significant difference compared to control. ‘#’ represented a significant difference in bystander effect w.r.t. irradiated and bystander cells (*p < 0.05, **p < 0.01, #p < 0.05, ##p < 0.01)
Phosphatidylserine (PS) externalization in bystander cells under the influence of γ-rays indicated increased cellular apoptosis (Fig. 3F–H). The increase was significant in 5 By and 8 By cells at different time points (Fig. 3F–H). 1 h ICCM treatment caused a dose-dependent increase in the percentage of bystander apoptotic cells, with 8 By cells showing the highest percentage of cells in the late apoptosis stage (Fig. 3F, I). The effects in 1 h ICCM treated 5 By, and 8 By cells were less than respective irradiation controls (Fig. 3I). 2 h ICCM induced a similar level of effects in 2 By and 5 By cells with significantly higher effects in 8 By cells (Fig. 3G). The effects in 24 h ICCM treated 5 By and 8 By cells were similar but significantly higher than 2 By cells (Fig. 3J). γ-rays at a dose of 2 Gy did not stimulate bystander apoptosis at any time post-irradiation (Fig. 3J). 8 By cells showed a time-dependent decrease in apoptosis with maximum effect in 1 h and minimum in 24 h ICCM treated cells (Fig. 3J). An increase in PS externalization was associated with increased cleaved PARP levels at both early and late time points in 8 By cells. Post 2 h ICCM treatment significant large positive relationship was also noticed with cleaved cas-3 and cas-9 (Fig. 3K).
Phosphorylation of H2AX had a significantly large positive relationship with the phospho-p53 level in γ-ray induced bystander HepG2 cells
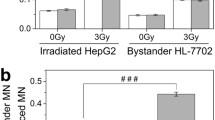
γ-rays increased H2AX phosphorylation and phospho-p53 levels in bystander cells which was confirmed by flow cytometry and western blotting (Fig. 4A–C, E). The increase was significant in all conditions other than 1 h and 2 h ICCM treated 2 By cells (Fig. 4A, F, G), which showed a similar response to irradiated cells. The effects in all other bystander cells were significantly lower than respective irradiation controls (Fig. 4A–C, E). Although 1 h ICCM treatment resulted in a radiation dose-dependent increase in p53 expression in bystander cells (Fig. 4G), phospho-H2AX level showed a dose-dependent increase only in 24 h ICCM treated cells (Fig. 4F). The 24 h ICCM treatment caused the most substantial increase in H2AX phosphorylation in bystander cells at different radiation doses (Fig. 4A, C, F) compared to the control group. Both phospho-H2AX and phospho-p53 demonstrated the highest increase in 24 h ICCM treated 8 By cells (Fig. 4F, G). Compared with 24 h ICCM treated 8 By BRL-3A cells, p53 level was 6% higher in HepG2 cells (Fig. 4E, I). However, there was no considerable difference in phospho-H2AX level between 8 By HepG2 and BRL-3A cells post 24 h ICCM treatment (Fig. 4D, H). In 8 By HepG2 cells, an increase in phospho-H2AX level was linked to increased phospho-p53 levels both in early and late time points post-irradiation (Fig. 4J). An increase in phospho-p53 level was also correlated with the increase in Bax levels (Fig. 4K). The increased phospho-p53 level was associated with a decreased phospho-Bcl2 level only in 24 h ICCM treated cells (Fig. 4L). In 2 h and 24 h ICCM treated 8 By HepG2 cells, phospho-H2AX level showed a significant large positive relationship with mitochondrial membrane depolarization (Fig. 4M).

Level of phosphorylated H2AX and p53 were modified in γ-ray induced bystander cells, showing the highest effects at the delayed time point. A–B The overlaid histograms represented the level of phospho-H2AX (A) and phospho-p53 (B) in the FL1 channel at different time points along the X-axis. The count was taken along the Y-axis. The shift in histogram towards the right indicated increased fluorescence intensity highlighting increased H2AX phosphorylation (A) and p53 phosphorylation (B). C–E The western blot images of phospho-H2AX (C, D) and phospho-p53 (E); C phospho-H2AX level in control, irradiated and bystander HepG2 cells post 24 h of irradiation and beta-Actin; D phospho-H2AX level in control and bystander BRL-3A cells and beta-Actin; E. phospho-p53 level in control, irradiated and bystander HepG2 cells and BRL-3A cells under different experimental conditions and beta-Actin. F The relative level of phospho-H2AX in bystander cells to control. G The relative level of phospho-p53 in bystander cells to loading control. H Comparative phospho-H2AX level in 24 h ICCM treated HepG2 and BRL-3A cells in cells. I Comparative phospho-p53 level in 24 h ICCM treated 8 By HepG2 and BRL-3A cells. J Significant large positive correlation between phospho-p53 and phospho-H2AX in 8 By HepG2 cells. K Substantial, large positive correlation between phospho-p53 and Bax in 8 By HepG2 cells. L Significant negative relationship between phospho-p53 and phospho Bcl2 in 24 h ICCM treated 8 By HepG2 cells. M Significant positive correlation between H2AX and mitochondrial membrane depolarization in 2 h and 24 h ICCM treated 8 By HepG2 cells. Values are mean ± S.E. ‘*’ represented significant difference compared to control. ‘#’ represented significant difference of bystander effect with respect to irradiated and bystander cells (*p < 0.05, **p < 0.01, #p < 0.05, ##p < 0.01)
A negative correlation was observed between pro-apoptotic Bax and anti-apoptotic phospho-Bcl2 levels in γ-ray induced bystander HepG2 cells
Compared with control cells, the expression of phospho-Bcl2 decreased (Fig. 5A–C, E). However, Bax expression increased in different ICCM treated bystander cells (Fig. 5D, E) except for 2 h ICCM treated 2 By and 5 By cells (Fig. 5H). Although 1 h ICCM caused a dose-dependent reduction in Bcl2 levels, the increase of Bax level demonstrated no dependency on the radiation dose (Fig. 5I, L). 2 h ICCM induced considerable effects on Bax and Bcl2 levels only in 8 By cells (Fig. 5E, J, L). In contrast, 24 h ICCM treated cells showed a dose-dependent increase in Bax levels, but the decrease in Bcl2 level was not dose-dependent (Fig. 5E, K, L). 8 By cells manifested the highest levels of Bax and the lowest levels of phospho-Bcl2 (Fig. 5E, K, L). While Bax showed a time-dependent increase with maximum effects in 24 h ICCM (Fig. 5M), effects on phospho-Bcl2 levels were maximum post 1 h ICCM treatment (Fig. 5L). Compared to BRL-3A cells under similar conditions, no substantial difference was observed (Fig. 5F, G, N). Compared to irradiated cells, Bax levels in the bystander HepG2 cells were significantly lower, except in the case of 2 By cells treated with 2 h ICCM (Fig. 5H). In contrast, phospho-Bcl2 levels in these bystander cells were similar to respective irradiation controls, except 2 h ICCM treated 2 By cells, and 1 h and 2 h ICCM treated 5 By cells, showing significantly higher levels (Fig. 5E). The ratio of Bax and Bcl2 in 8 By HepG2 cells was highest at 1 h but not significantly different from 24 h ICCM treatment (Fig. 5O), indicating bystander cellular susceptibility to apoptosis at both early and delayed time points post-irradiation. A correlation study pointed out that an increase in Bax levels in the bystander cells was associated with a decrease in Bcl2 levels (Fig. 5P). Although a significantly large positive relationship was noticed between Bax and phospho-H2AX, Bcl2 and phospho-H2AX showed a significant negative relationship (Fig. 5Q, R).

γ-radiation decreased phospho-Bcl2 level but increased Bax level in bystander HepG2 cells. A–D The overlaid histogram plots represented the level of A–C phospho-Bcl2 and D Bax, in FL1-H along the X-axis. The count was taken along Y-axis. A shift in histogram towards the left indicated decreased fluorescence intensity (A–C), indicating a decreased level of Bcl2 in A 1 h ICCM, B 2 h ICCM, C 24 h ICCM compared to control. A shift in histogram towards the right indicated increased fluorescence intensity (D), suggesting an increased level of Bax. E The western blot images of phospho-Bcl2 and Bax in control, irradiated, and bystander HepG2 cells under different experimental conditions and beta-Actin. F and G The western blot images of Bax (F) and phospho-Bcl2 (G) in control and bystander BRL-3A cells and beta-Actin. H–K Bax level in bystander HepG2 cells receiving H, J 2 h ICCM; I 1 h ICCM; K 24 h ICCM. L Bar diagram represented the relative level of phospho-Bcl2 in bystander HepG2 cells to control. M Percentage change in Bax level in 8 By HepG2 cells according to ICCM transfer time. N Relative phospho-Bcl2 and Bax levels in bystander BRL-3A and HepG2 cells in 1 h ICCM and 24 h ICCM treated 8 By cells, w.r.t. control. O Ratio of Bax and Bcl2 in 8 By HepG2 cells receiving 1 h and 24 h ICCM. P Significant negative relationship between Bax and phospho-Bcl2 in 8 By HepG2 cells. Q Significant positive correlation between BAX and phospho-H2AX. R Significant negative correlation between phospho-Bcl2 and phospho-H2AX. Values are mean ± S.E. ‘*’ represented significant difference compared to control. ‘#’ represented a significant difference in bystander effect compared to irradiated and bystander cells. (*p < 0.05, **p < 0.01, #p < 0.05, ##p < 0.01)
Increased cleaved cas-3 levels in bystander HepG2 cells was linked with increased cleaved cas-9 levels at all time points and with increased cleaved PARP levels only at earlier time points post γ-irradiation
Transfer of ICCM at different times post-irradiation increased cleaved cas-3, cleaved cas-9, and cleaved-PARP in the bystander cells (Fig. 6A–F). In 2 By cells, no considerable increase in cleaved cas-9 level could be observed at any time point, but 1 h ICCM treatment caused an increase in cleaved cas-3 and cleaved PARP levels (Fig. 6A–C). Although cleaved cas-9 level in 2 By cells was similar to respective irradiation controls, 5 By and 8 By cells showed significantly lower levels than irradiated cells at all time points (Fig. 6A, D). Except for 2 h ICCM treated 2 By cells, and 24 h ICCM treated 5 By and 8 By cells, cleaved cas-3 levels in the bystander cells were also significantly lower than the irradiated cells (Fig. 6B, E, H). The bystander cells also showed lower cleaved PARP levels than irradiated cells, except 2 h ICCM, treated 2 By and 8 By cells and 24 h ICCM treated 2 By cells showing similar levels with irradiated cells (Fig. 6C, F, J). In 5 By and 8 By cells, the effects induced by 1 h and 2 h ICCM were similar but significantly higher than 24 h ICCM. While this was true for cleaved cas-3 and cleaved cas-9, in 8 By cells cleaved PARP level was similar post 1 h, 2 h, or 24 h ICCM treatment (Fig. 6H–J). 1 h ICCM treated 5 By cells and 2 h ICCM treated 8 By cells showed a similar degree of effects and maximum cleaved PARP level among all the experimental groups (Fig. 6J). The highest effects on the cleaved cas-9 level were seen in 1 h ICCM treated 8 By cells, but this was not significantly different from 2 h ICCM treated 5 By and 8 By cells (Fig. 6I). The highest levels of cleaved cas-3 were noted in 1 h and 2 h ICCM treated 8 By cells (Fig. 6H). The effects in 8 By HepG2 cells were significantly lower than 8 By BRL-3A cells when treated with the same ICCM (Fig. 6K). At all time points, an increase in cleaved cas-3 in 8 By cells was correlated to a rise in cleaved cas-9 level (Fig. 6L). At the earlier time points of 1 h or 2 h post-irradiation, increased cleaved cas-3 level in bystander cells was positively linked to increased cleaved PARP levels. However, no relationship could be noticed between cas-9 and cleaved PARP levels (Fig. 8A–C).

γ-rays increased the cleavage of cas-9, cas-3, and PARP in bystander cells. A, C The overlaid histogram plots represented the level of A cleaved cas-9 and C cleaved PARP in FL1-H along the X-axis. The count was taken along Y-axis. A shift in histogram towards the right indicated increased fluorescence intensity. Compared to control, the increased histogram shift indicated an increased level of cleaved A. cas-9, C PARP. B The histogram plots represented the level of cleaved cas-3 in control, irradiated, and bystander cells post 1 h of irradiation. Cleaved cas-3 was taken in FL1-H along the X-axis and count along the Y-axis. D–G The western blot images of D. cleaved cas-9 in control, irradiated and bystander HepG2 and BRL-3A cells at different time points post-irradiation and β-actin; E cleaved cas-3 in control, irradiated and bystander HepG2 and BRL-3A cells at 1 h post-irradiation and β-actin; F cleaved PARP in control, irradiated and bystander HepG2 cells at different time points post-irradiation and β-actin; G cleaved PARP in control, and bystander BRL-3A cells at different time points post-irradiation and β-actin. H The time-dependent change in the level of cas-3 in different bystander cells w.r.t. control. I The relative level of cleaved cas-9 in bystander cells to control. J The change in the level of cleaved PARP in 2 By, 5 By, 8 By cells w.r.t. control post 1 h, 2 h, and 24 h ICCM treatment. K Relative levels of cleaved cas-3, cleaved cas-9, and cleaved PARP in 8 By HepG2 and BRL-3A cells to control. L Significant large positive correlation between cleaved cas-3 and cleaved cas-9. Values are mean ± S.E. ‘*’ represented significant difference compared to control. ‘#’ represented a significant difference in bystander effect compared to irradiated and bystander cells (*p < 0.05, **p < 0.01, #p < 0.05, ##p < 0.01)
SEM studies outlined distinct modifications in cellular morphology
Scanning electron microscopic studies showed distinct morphological alterations in bystander cells compared to control (Fig. 7E–M). Control cells (Fig. 7A) were slightly flattened and attached to the substratum; microvilli were scanty and, when present, distributed along the margins. The cell surface was homogenous, cell symmetrical. Distinct filopodia were present, and there was no membrane blebbing. Exposure of cells to γ-rays induced alterations in cell shape with a marked increase in the number of microvilli (Fig. 7B–D). Cells demonstrated asymmetry and cell surface heterogeneity. At a dose of 8 Gy, cells were distorted, shriveled with damaged filopodia, highly increased number of microvilli, and membrane blebbing (Fig. 7D). Bystander cells at 1 h post-irradiation showed maximum cell surface alterations (Fig. 7E–G). At a dose of 8 Gy, 1 h ICCM treated bystander cells became rounded with cell surface distortions and a few membrane blebs (Fig. 7G). The radiation dose and the time of ICCM transfer affected cell morphology and surface topography.

SEM studies in bystander cells. A SEM image of control HepG2 cells. B–D SEM images of HepG2 cells 24 h post-irradiation with different doses of γ-rays. E–G SEM images of bystander HepG2 cells at different γ-ray doses post 1 h of irradiation. H–J SEM images of bystander HepG2 cells at different γ-ray doses post 2 h of irradiation. K–M SEM images of bystander HepG2 cells at different γ-ray doses post 24 h of irradiation
Discussion
Previous studies have shown that ionizing radiation induces bystander cell apoptosis, especially the intrinsic pathway of apoptosis [2, 15,16,17,18,19]. During clinical radiotherapy, RIBE may be beneficial by promoting cancer cell death but may increase secondary cancer risks by affecting normal cells. Hence, our study investigated the effect of γ-irradiated HepG2 cells on bystander HepG2 cells compared to bystander Brl-3a cells to evaluate the underlying signaling cross-talk promoting bystander cellular death. The results showed that: (1) γ-rays activate the intrinsic pathway of apoptosis in bystander HepG2 cells, (2) the release of bystander signals from irradiated cells and subsequent effects on bystander cells depends on the radiation dose, (3) the time of ICCM transfer post-γ-irradiation affect the development of bystander responses, (4) the release of bystander signals from irradiated cells is persistent up to 24 h, and most importantly, (5) DNA damage markers, mitochondrial membrane potential, and apoptosis markers cross-talk with each other to shape bystander HepG2 cell fate under the influence of γ-rays.
Phosphorylation of the histone variant H2AX is a marker for DNA damage [31]. Ionizing radiation exposure induces H2AX phosphorylation at Ser 139 (γ-H2AX) at sites of DNA damage [32, 33]. In addition to activating DNA damage response pathways, H2AX phosphorylation promotes apoptosis by activating DNA fragmentation [34]. The tumor suppressor protein p53 plays a vital role in the cellular response to DNA damage. DNA damage induces p53 phosphorylation at Ser15. Existing literature has reported the development of γ-H2AX foci in various bystander cell types [35,36,37] and the activation of p53 in bystander cells [38,39,40,41,42]. In our study, increased phosphorylated H2AX indicated DNA damage, especially DNA double-stranded break formation in the radioresistant bystander HepG2 cells. According to Sedelnikova et al. (2007), DSBs in bystander cells reach maximum levels from 12 to 48 h post-irradiation in human tissues [43]. Our results also showed that out of the three time points examined (1 h, 2 h, and 24 h), 24 h ICCM had the highest effect on the phospho-H2AX level. We also observed the highest p53 phosphorylation in bystander cells at 24 h post-irradiation. Our observation was similar to the reports of Korturbash et al. (2008) and Hamada et al. (2008), who suggested that bystander cells manifest delayed p53 phosphorylation [44, 45]. 8 Gy dose induced the maximum phosphorylation of H2AX and p53 in bystander cells. Pereira et al. (2014) also reported the dose-dependency of γ-H2AX foci in bystander zebrafish cells [46]. Tomita et al. (2013) pointed out the regulation of effects at different doses by the p53 status in bystander cells [47]. However, our results were contradictory to Hu et al. (2006), who reported early observation of DSB (maximum at 30 min post-irradiation) and dose independence in bystander cells [48]. This disparity in observation was possibly due to the difference in cell types and linear energy transfer (LET) of radiation. Hu et al. (2006) studied normal skin fibroblasts, which have different sensitivity to bystander signals than tumor cells. In addition, the energy deposition from high LET radiation like ɑ-particles used in their study can lead to larger local doses, which trigger gene expression patterns, different from what is seen after exposure to low LET radiation like γ-rays at similar doses [49,50,51]. In our study, at all time points post-irradiation, bystander HepG2 cells showed a similar trend in the levels of H2AX and p53 phosphorylation (Fig. 8A–C). This indicated that ICCM borne factors from irradiated cells induced DNA damage, activating p53 molecules in the bystander cells. At 24 h post-irradiation, increased phospho-H2AX and phospho-p53 were linked to decreased anti-apoptotic Bcl2 levels (Fig. 8C). The positive correlation between phospho-p53 and Bax levels at all time points confirmed the role of p53 in bystander apoptosis by regulating Bax molecules (Fig. 8A–C). Widel et al. (2015), however opined that p53 null bystander cells show higher apoptosis. However, their radiation source was X-rays, and they studied effects on colorectal carcinoma cells [52].

Correlation matrix showing the correlation between different parameters studied in 8 By HepG2 cells at A 1 h, B 2 h, and C 24 h post-γ-irradiation. Relationships that were not statistically significant were marked with the ‘X’ sign. Different colors indicate different r-values showing positive and negative correlations
Mitochondria are the essential controller of cellular function and metabolism. Mitochondria regulate redox homeostasis, maintain energy (ATP) production through oxidative phosphorylation, modulate redox signaling by producing reactive oxygen species (ROS), and control cell death through apoptosis. In addition to these critical cell functions, recent evidence supports a signaling role for mitochondria. In viable cells, mitochondrial metabolic enzymes can transform the MTT tetrazolium salt into a deep purple-colored product, MTT formazan. Ionizing γ rays interfere with cell metabolic enzyme functioning, so irradiated cell viability decrease compared to unirradiated control. Mitochondrial membrane potential is a crucial determining factor of mitochondrial function. The depolarization of the mitochondrial membrane is an important feature in the early stage of apoptosis. From our studies, it can be concluded that γ rays alter mitochondrial activity in terms of enzyme functioning and membrane potential in both irradiated and bystander cells. We observed decreased viability of bystander cells at different radiation doses with the most significant impact on 8 By cells. Previous studies have pointed out the dose-dependency of bystander cell viability [52,53,54,55]. In our study, the viability of bystander cells at different doses was higher than irradiated cells, similar to observations of Faqihi et al. (2015) [19]. Our results complied with the observations of Wu et al. (2011) and Rezaei et al. (2021) [23, 56] because ICCM treatment had no significant effect on bystander cells at different time points post-irradiation. However, Wu et al. (2011) reported an increased survival of bystander HepG2 under the influence of high LET carbon ions, contrary to our observations [23]. Although the cell line was the same, the observed differences were due to the differences in LET. Different studies emphasize that LET is an important regulator of bystander effects, and the difference of bystander effect of the same cell type is a function of LET [49, 57,58,59]. The LET of ionizing radiation modulates the relative biological effectiveness (RBE) of the exposed cells. Some reports [58, 60] speculate that radiation quality causes differential activation of signaling pathways in the targeted cells, stimulating the release of LET-specific bystander signals from the same cell. As evident from JC-1 staining experiments, we observed the most considerable impact on mitochondrial membrane potential in 2 h ICCM treated 5 By cells and in 8 By cells, whose effects were independent of time. In 2 By and 5 By cells, effects increased post 2 h ICCM treatment and decreased after 24 h. Contrary to the observation of Lyng et al. (2000), we observed a significant increase in mitochondrial depolarization in bystander cells treated with ICCM at all time points [24]. The difference in observation can be attributed to the differential sensitivity of normal keratinocytes and tumor cells to bystander signals. The study of this group emphasized that γ-ray induced effect in bystander keratinocyte was independent of dose, which was partly in compliance with our observation because 2 h ICCM treatment caused no significant difference in effects in different doses [24]. In our study, a positive association was noticed between mitochondrial membrane depolarization and pro-apoptotic Bax at all time points post-irradiation (Fig. 8A–C). Since Bax level was also linked with phospho-p53 levels, it is evident from our observation that p53 regulates Bax level while Bax regulates mitochondrial membrane depolarization. At 2 h and 24 h post-irradiation, increased correlations between phospho-H2AX and mitochondrial membrane depolarization indicated the involvement of oxidative stress factors like ROS in the induction of bystander cell damage (Fig. 8B, C).
Our study showed that under the influence of γ-rays, the levels of apoptosis markers, such as Bax, cleaved cas-9, cleaved cas-3, and cleaved PARP in bystander HepG2 cells significantly increased, while Bcl2 phosphorylation decreased. The phosphatidylserine externalization studies also indicated γ-ray-induced bystander apoptosis. We observed the highest Bax level at 24 h post-irradiation while pro-survival Bcl2 showed a maximum increase at 1 h post-incubation in bystander cells. Previous studies in the existing literature have also pointed out that in Bax, expression is highest at the delayed time points in radioresistant cell lines. In contrast, Bcl2 expression is maximum at early time (1 h) post-γ-irradiation [25, 26]. The cleaved cas-3, cas-9, and PARP levels reached their maximum at the earlier time points (1 h, 2 h). Therefore, radiation exposure has diverse effects on apoptotic markers in bystander cells at discrete time points. These results were similar to the observations of Furlong et al. (2013) [2]. In our study, PI-Annexin staining of cells confirmed that apoptosis in bystander HepG2 cells was maximum at 1 h post γ irradiation. Chen et al. (2020) also reported increased PS externalization in bystander cells [61]. In contrast, Fu et al. (2016) reported that radiosensitive bystander cells showed increased apoptosis at 24 h and reached the highest effect at 48 h [62]. However, in bystander lung cancer cells, high dose (8 Gy) ICCM decreased apoptosis compared to control at 72 h post-irradiation. Yoshino et al. speculated in 2019 that bystander signals (such as cytokines) are involved in increasing radio-resistance of bystander cells at delayed times [63], which may be responsible for time-dependent variations of apoptotic markers in different cell types [16]. Li et al. (2017) also reported a similar dose-dependent increase in bystander apoptosis in an in vivo study [22]. In bystander erythroleukemia cells, γ-rays activated Bax, Bcl-2, and cleavage of poly (ADP-ribose) polymerase [25]. The study by Zhang et al. (2012) emphasized the effect of γ-rays on the Bcl-2/Bax ratio in HepG2 cells, which decreased with the time of treatment of conditioned media [26]. In our study, the Bax/Bcl2 ratio was highest in 1 h ICCM treated 8 By cells, but significantly higher than control post 24 h ICCM treatment. This indicated persistent susceptibility of bystander cells to apoptosis, although the maximum apoptosis was observed in 1 h ICCM receiving cells. The effects of γ-rays on the gene expression of apoptosis markers in bystander HaCaT keratinocytes were similar to our observations. El din et al. (2017) noted an increase in caspase-3 levels in bystander cells [64]. Cranial irradiation of rats induced apoptotic cell death with decreased Bcl-2, increased p53, Bax, cas-3, and DNA damage in bystander spleen [64]. We observed that the 8 Gy dose of γ-ray had the maximum effects on Bax, phospho-Bcl2, cleaved cas-3, and cas-9 in bystander cells. Furlong et al. also emphasized the dose-dependent expression of Bax, Bcl2, and caspase-3 in their 2013 study [2]. SEM studies showed distinct morphological alterations in bystander cells, with the most considerable effects in 1 h ICCM treated 8 By cells. At 1 h and 24 h post-irradiation, pro-apoptotic Bax showed negative regulation of anti-apoptotic phospho Bcl2 levels (Fig. 8A, C). At the earlier time points (1 h, 2 h) post-irradiation, mitochondrial membrane depolarization positively regulated caspase-9 cleavage (Fig. 8A, B). Since no correlation was observed between cleaved cas-3 levels and mitochondrial membrane depolarization, but cleaved cas-9 and cas-3 levels showed a positive association, we conclude that cleaved cas-9 regulated caspase-3 cleavage at all time points (Fig. 8A–C). The link between PARP cleavage with only cleaved cas-3 and not cleaved cas-9 suggest that cas-3 regulates PARP cleavage (Fig. 8A, B). PARP cleavage, the marker of cellular apoptosis, manifested a positive relationship with PS externalization at all the time points studied (Fig. 8A–C). Besides these key interactions observed between DNA damage markers and apoptosis markers in the bystander cells, some specific interactions were observed at particular time points post-irradiation. At 1 h post-irradiation, negative correlations were observed between cellular viability and apoptosis markers like PS externalization, cas-3, and PARP cleavage (Fig. 8A). A decrease in phospho-Bcl2 levels was correlated with increased mitochondrial membrane depolarization (Fig. 8A). In 2 h ICCM treated cells, decreased cell viability was linked with increased activation of DNA damage markers, H2AX and p53, increased mitochondrial membrane depolarization, and Bax levels (Fig. 8B). A positive correlation was noted between PS externalization and cleaved cas-3 and cas-9 levels. DNA damage marker H2AX showed a positive association with apoptosis regulators like Bax and cleaved Cas-9 levels. An increase in phospho-p53 and mitochondrial membrane depolarization followed a similar trend (Fig. 8B).
The data from the existing literature have indicated the regulatory influence of different factors, such as cell/tissue specificity, linear energies, and radiation dose, on the radiation-induced bystander effects [4, 65]. Although bystander effects are absent between irradiated lung cancer and bystander fibroblasts, irradiated skin cancer cells induce dose-dependent effects on bystander skin fibroblasts [66, 67]. ICCM from high LET iron ion irradiated skin fibroblasts induce higher effects on these bystander cells than low LET protons [49, 57, 66, 67]. However, ICCM from low LET X-ray irradiated prostate cancer cells induces higher effects on bystander skin fibroblasts than high LET α-particles or carbon ions [58, 59]. It has been understood that bystander effects in each cell type are unique due to differential activation of response pathways when exposed to different radiation sources. Thus, the study of bystander effects is necessary for different cell lines under different radiation sources and time points to understand the diversity of responses. Our study is the first to highlight γ-ray-induced effects at three different doses and three time points post-irradiation to have a complete idea about the interactions of signaling mediators in bystander cells that regulate response. The post-irradiation time is an important factor that affects the expression of the signaling molecules in bystander cells differently. Although the highest level of DNA damage was observed 24 h post-irradiation, apoptosis was maximum at early time post-irradiation. This suggests that apoptosis in bystander cells might be independent of DNA damage and induced directly by the bystander stress signals from irradiated cells. Additional stress factors (like ROS generation, oxidative stress), which have not been explored in our study, may be possible mediators of apoptosis at the early time points. Nevertheless, our study showed persistence of DNA damage in bystander cells from an early time point (1 h), indicating the possible involvement of both DNA damage and additional stress factors in inducing apoptosis. The main conclusions regarding the interactions between the different signaling mediators in bystander HepG2 cells, specifically DNA damage markers and apoptosis regulators, have been represented in Fig. 9 to highlight the main findings of our study.

γ-radiation causes activation of different signaling mediators in bystander cells stimulating DNA damage and apoptosis. This figure was created using images from Servier Medical Art Commons Attribution 3.0 Unported License. (http://smart.servier.com). Servier Medical Art by Servier is licensed under a Creative Commons Attribution 3.0 Unported License
Conclusion
This study focused on the effects of γ-irradiation on bystander HepG2 and Brl3a cells to understand the underlying intracellular signaling cross-talk in bystander liver cells. Although many studies [5, 23, 26, 39, 68,69,70,71,72] focused on the bystander effects of γ-rays on HepG2 cells, the experimental conditions and the parameters probed in our research were different. In some studies, where some similar parameters were explored, the cell line and the radiation sources were different [2, 22, 24, 25, 35,36,37, 42, 44, 54,55,56, 61, 62, 64]. The novelty of our research work lies in elucidating the effects of γ-irradiated HepG2 ICCM on bystander HepG2 and Brl-3a cells at 3 different doses and time points for explicit understanding of the scenario in a comparative manner. We analyzed the status of apoptosis and DNA damage markers, quantified bystander cell death using PI-Annexin assay, and examined morphological changes of bystander cells. The main advantage of this work is that it has highlighted the correlations between the different parameters studied at different time points post-irradiation, which have not been included in any of the previous studies.
Our results showed that the γ-radiation dose affects the activation of different apoptosis and DNA damage markers in bystander HepG2 cells. This study focuses on the signaling cross-talk that regulates bystander cellular apoptosis providing a complete idea about their interactions and correlations. DNA damage markers and apoptosis regulators work in synergy to induce bystander cell death. The release of the bystander signals is persistent at 24 h in all parameters studied though the intensity of the signal showed time-dependent variations. We cultured bystander cells simultaneously (1 h) in ICCM collected at different time points after γ-irradiation. Therefore, the variation in observations may not be due to specific reactions in bystander cells. Post 24 h, γ-irradiated HepG2 cells released factors that actuated the highest level of DNA damage, p53 activation, and Bax level in bystander cells. However, post early time points of 1 h or 2 h, factors released from the irradiated cells induced maximum activation of pro-apoptotic factors in bystander cells. This was confirmed by PI-Annexin and SEM studies, which showed the highest bystander apoptosis in the conditioned media, collected 1 h post-irradiation. It can be concluded that the time-specific release of signaling mediators from irradiated HepG2 cells causes differential activation of signaling molecules in bystander cells. The study identified the radiation dose and time point exerting the highest effects on different signaling mediators in bystander HepG2 cells, and the effects were compared with normal bystander Brl-3a cells to understand the overall tissue response to radiation. We performed the studies up to 24 h, but in the future, bystander effects of γ rays on HepG2 cells need to be analyzed 48–72 h post-irradiation. The diversity of observation warrants further investigations to reveal the exact mechanisms of various bystander responses.
Data availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Mavragani IV, Nikitaki Z, Kalospyros SA, Georgakilas AG (2019) Ionizing radiation and complex DNA damage: from prediction to detection challenges and biological significance. Cancers (Basel) 11:1789. https://doi.org/10.3390/cancers11111789
Furlong H, Mothersill C, Lyng FM, Howe O (2013) Apoptosis is signalled early by low doses of ionizing radiation in a radiation-induced bystander effect. Mutat Res 741–742:35–43. https://doi.org/10.1016/j.mrfmmm.2013.02.001
Gallucci S, Matzinger P (2001) Danger signals: SOS to the immune system. Curr Opin Immunol 13:114–119. https://doi.org/10.1016/S0952-7915(00)00191-6
Mukherjee S, Chakraborty A (2019) Radiation-induced bystander phenomenon: insight and implications in radiotherapy. Int J Radiat Biol 95:243–263. https://doi.org/10.1080/09553002.2019.1547440
He M, Zhao M, Shen B, Prise KM, Shao C (2011) Radiation-induced intercellular signaling mediated by cytochrome-c via a p53-dependent pathway in hepatoma cells. Oncogene 30:1947–1955. https://doi.org/10.1038/onc.2010.567
Cao X, Wen P, Fu Y, Gao Y, Qi X, Chen B, Tao Y, Wu L, Xu A, Lu H, Zhao G (2019) Radiation induces apoptosis primarily through the intrinsic pathway in mammalian cells. Cell Signal 62:109337. https://doi.org/10.1016/j.cellsig.2019.06.002
Loewer A, Karanam K, Mock C, Lahav G (2013) The p53 response in single cells is linearly correlated to the number of DNA breaks without a distinct threshold. BMC Biol 11:114. https://doi.org/10.1186/1741-7007-11-114
Sia J, Szmyd R, Hau E, Gee HE (2020) Molecular mechanisms of radiation-induced cancer cell death: a primer. Front Cell Dev Biol 8:41. https://doi.org/10.3389/fcell.2020.00041
Maier P, Hartmann L, Wenz F, Herskind C (2016) Cellular pathways in response to ionizing radiation and their targetability for tumor radiosensitization. Int J Mol Sci 17:102. https://doi.org/10.3390/ijms17010102
Walsh DWM, Siebenwirth C, Greubel C, Ilicic K, Reindl J, Girst S, Muggiolu G, Simon M, Barberet P, Seznec H, Zischka H, Multhoff G, Schmid TE, Dollinger G (2017) Live cell imaging of mitochondria following targeted irradiation in situ reveals rapid and highly localized loss of membrane potential. Sci Rep 7:46684. https://doi.org/10.1038/srep46684
Smaili SS, Hsu YT, Sanders KM, Russell JT, Youle RJ (2001) Bax translocation to mitochondria subsequent to a rapid loss of mitochondrial membrane potential. Cell Death Differ 8:909–920. https://doi.org/10.1038/sj.cdd.4400889
Gómez-Crisóstomo NP, López-Marure R, Zapata E, Zazueta C, Martínez-Abundis E (2013) Bax induces cytochrome c release by multiple mechanisms in mitochondria from MCF7 cells. J Bioenerg Biomembr 45:441–448. https://doi.org/10.1007/s10863-013-9508-x
Chang HY, Yang X (2000) Proteases for cell suicide: functions and regulation of caspases. Microbiol Mol Biol Rev 64:821–846. https://doi.org/10.1128/mmbr.64.4.821-846.2000
Eriksson D, Löfroth PO, Johansson L, Riklund K, Stigbrand T (2009) Apoptotic signalling in HeLa Hep2 cells following 5 Gy of cobalt-60 gamma radiation. Anticancer Res 29:4361–4366
Lyng FM, Seymour CB, Mothersill C (2002) Initiation of apoptosis in cells exposed to medium from the progeny of irradiated cells: a possible mechanism for bystanderinduced genomic instability? Rad Res 157:365–370. https://doi.org/10.1667/0033-7587(2002)157[0365:ioaice]2.0.co;2
Lyng FM, Maguire P, Kilmurray N, Mothersill C, Shao C, Folkard M, Prise KM (2006) Apoptosis is initiated in human keratinocytes exposed to signalling factors from microbeam irradiated cells. Int J Radiat Biol 82:393–399. https://doi.org/10.1080/09553000600803904
Maguire P, Mothersill C, Seymour C, Lyng FM (2005) Medium from irradiated cells induces dose-dependent mitochondrial changes and BCL2 responses in unirradiated human keratinocytes. Radiat Res 163:384–390. https://doi.org/10.1667/rr3325
He G, Tang A, Xie M, Xia W, Zhao P, Wei J, Lai Y, Tang X, Zou YM, Liu H (2020) Blood gene expression profile study revealed the activation of apoptosis and p53 signaling pathway may be the potential molecular mechanisms of ionizing radiation damage and radiation-induced bystander effects. Dose Response 18(1):1559325820914184. https://doi.org/10.1177/1559325820914184
Faqihi F, Neshastehriz A, Soleymanifard S, Shabani R, Eivazzadeh N (2015) Radiation-induced bystander effect in non-irradiated glioblastoma spheroid cells. J Radiat Res 56:777–783. https://doi.org/10.1093/jrr/rrv039
Seymour CB, Mothersill C (2000) Relative contribution of bystander and targeted cell killing to the low-dose region of the radiation dose-response curve. Radiat Res 153:508–511. https://doi.org/10.1667/0033-7587(2000)153[0508:rcobat]2.0.co;2
Gow MD, Seymour CB, Byun SH, Mothersill CE (2008) Effect of dose rate on the radiation-induced bystander response. Phys Med Biol 53:119–132. https://doi.org/10.1088/0031-9155/53/1/008
Li Q, Shi J, Chen L, Zhan F, Yuan H, Wang J, Xu A, Wu L (2017) Spatial function of the oxidative DNA damage response in radiation induced bystander effects in intra- and inter-system of Caenorhabditis elegans. Oncotarget 8:51253–51263. https://doi.org/10.18632/oncotarget.17229
Wu Q, Li Q, Jin X, Liu X, Dai Z (2011) Bystander effect in human hepatoma HepG2 cells caused by medium transfers at different times after high-LET carbon ion irradiation. Nucl Instrum Methods Phys Res B 269:153–158. https://doi.org/10.1016/j.nimb.2010.10.012
Lyng FM, Seymour CB, Mothersill C (2000) Production of a signal by irradiated cells which leads to a response in unirradiated cells characteristic of initiation of apoptosis. Br J Cancer 83:1223–1230. https://doi.org/10.1054/bjoc.2000.1433
Mitra AK, Krishna M (2007) Radiation-induced bystander effect: activation of signaling molecules in K562 erythroleukemia cells. J Cell Biochem 100:991–997. https://doi.org/10.1002/jcb.21084
Zhang J, Xie Y, Xu Y, Shao C (2012) Suppression of endogenous hydrogen sulfide contributes to the radiation-induced bystander effects on hypoxic HepG2 cells. Radiat Res 178:395–402. https://doi.org/10.1667/RR2967.1
Yang H, Anzenberg V, Held KD (2007) The time dependence of bystander responses induced by iron-ion radiation in normal human skin fibroblasts. Radiat Res 168:292–298
Harada K, Nonaka T, Hamada N, Sakurai H, Hasegawa M, Funayama T, Kakizaki T, Kobayashi Y, Nakano T (2009) Heavy-ion-induced bystander killing of human lung cancer cells: role of gap junctional intercellular communication. Cancer Sci 100:684–688
He M, Dong C, Ren R, Yuan D, Xie Y, Pan Y, Shao C (2013) Radiation enhances the invasiveness of irradiated and nonirradiated bystander hepatoma cells through a VEGF-MMP2 pathway initiated by p53. Radiat Res 180:389–397
Przybyszewski WM, Widel M, Szurko A, Lubecka B, Matulewicz L, Maniakowski Z, Polaniak R, Birkner E, Rzeszowska-Wolny J (2004) Multiple bystander effect of irradiated megacolonies of melanoma cells on non-irradiated neighbours. Cancer Lett 214:91–102
Mah LJ, El-Osta A, Karagiannis TC (2010) γH2AX: a sensitive molecular marker of DNA damage and repair. Leukemia 24:679–686. https://doi.org/10.1038/leu.2010.6
Rogakou EP, Boon C, Redon C, Bonner WM (1999) Megabase chromatin domains involved in DNA double-strand breaks in vivo. J Cell Biol 146:905–916. https://doi.org/10.1083/jcb.146.5.905
Redon CE, Dickey JS, Bonner WM, Sedelnikova OA (2009) γ-H2AX as a biomarker of DNA damage induced by ionizing radiation in human peripheral blood lymphocytes and artificial skin. Adv Space Res 43:1171–1178. https://doi.org/10.1016/j.asr.2008.10.011
Lu C, Zhu F, Cho YY, Tang F, Zykova T, Ma W, Bode AM, Dong Z (2006) Cell apoptosis: requirement of H2AX in DNA ladder formation but not for the activation of caspase-3. Mol Cell 23:121–132. https://doi.org/10.1016/j.molcel.2006.05.023
Sokolov MV, Smilenov LB, Hall EJ, Panyutin IG, Bonner WM, Sedelnikova OA (2005) Ionizing radiation induces DNA double-strand breaks in bystander primary human fibroblasts. Oncogene 24:7257–7265. https://doi.org/10.1038/sj.onc.1208886
Dickey JS, Baird BJ, Redon CE, Sokolov MV, Sedelnikova OA, Bonner WM (2009) Intercellular communication of cellular stress monitored by γ-H2AX induction. Carcinogenesis 30:1686–1695. https://doi.org/10.1093/carcin/bgp192
Burdak-Rothkamm S, Short SC, Folkard M, Rothkamm K, Prise KM (2007) ATR-dependent radiation-induced gamma H2AX foci in bystander primary human astrocytes and glioma cells. Oncogene 26:993–1002. https://doi.org/10.1038/sj.onc.1209863
Jasty R, Lu J, Irwin T, Suchard S, Clarke MF, Castle VP (1998) Role of p53 in the regulation of irradiation-induced apoptosis in neuroblastoma cells. Mol Genet Metab 65:155–164. https://doi.org/10.1093/10.1006/mgme.1998.2747
Li J, He M, Shen B, Yuan D, Shao C (2013) Alpha particle-induced bystander effect is mediated by ROS via a p53-dependent SCO2 pathway in hepatoma cells. Int J Radiat Biol 89:1028–1034. https://doi.org/10.3109/09553002.2013.817706
Mothersill C, Bristow RG, Harding SM, Smith RW, Mersov A, Seymour CB (2011) A role for p53 in the response of bystander cells to receipt of medium borne signals from irradiated cells. Int J Radiat Biol 87:1120–1125. https://doi.org/10.3109/09553002.2011.610866
He M, Dong C, Xie Y, Li J, Yuan D, Bai Y, Shao C (2014) Reciprocal bystander effect between ɑ-irradiated macrophage and hepatocyte is mediated by cAMP through a membrane signaling pathway. Mutat Res 763–764:1–9. https://doi.org/10.1016/j.mrfmmm.2014.03.001
He M, Dong C, Konishi T, Tu W, Liu W, Shiomi N, Kobayashi A, Uchihori Y, Furusawa Y, Hei TK, Dang B, Shao C (2014) Differential effects of p53 on bystander phenotypes induced by gamma ray and high LET heavy ion radiation. Life Sci Space Res (Amst) 1:53–59. https://doi.org/10.1016/j.lssr.2014.02.003
Sedelnikova OA, Nakamura A, Kovalchuk O, Koturbash I, Mitchell SA, Marino SA, Brenner DJ, Bonner WM (2007) DNA double-strand breaks form in bystander cells after microbeam irradiation of three-dimensional human tissue models. Cancer Res 67:4295–4302. https://doi.org/10.1158/0008-5472.CAN-06-4442
Koturbash I, Loree J, Kutanzi K, Koganow C, Pogribny I, Kovalchuk O (2008) In vivo bystander effect: cranial X-irradiation leads to elevated DNA damage, altered cellular proliferation and apoptosis, and increased p53 levels in shielded spleen. Int J Radiat Oncol Biol Phys 70:554–562. https://doi.org/10.1016/j.ijrobp.2007.09.039
Hamada N, Ni M, Funayama T, Sakashita T, Kobayashi Y (2008) Temporally distinct response of irradiated normal human fibroblasts and their bystander cells to energetic heavy ions. Mutat Res 639(1–2):35–44. https://doi.org/10.1016/j.mrfmmm.2007.11.001
Pereira S, Malard V, Ravanat JL, Davin AH, Armengaud J, Foray N, Adam-Guillermin C (2014) Low doses of gamma-irradiation induce an early bystander effect in zebrafish cells which is sufficient to radioprotect cells. PLoS ONE 9:e92974. https://doi.org/10.1371/journal.pone.0092974
Tomita M, Maeda M, Kobayashi K, Matsumoto H (2013) Dose response of soft X-ray-induced bystander cell killing affected by p53 status. Radiat Res 179:200–207. https://doi.org/10.1667/RR3010.1
Hu B, Wu L, Han W, Zhang L, Chen S, Xu A, Hei TK, Yu Z (2006) The time and spatial effects of bystander response in mammalian cells induced by low dose radiation. Carcinogenesis 27:245–251. https://doi.org/10.1093/carcin/bgi224
Buonanno M, de Toledo SM, Pain D, Azzam EI (2011) Long-term consequences of radiation-induced bystander effects depend on radiation quality and dose and correlate with oxidative stress. Radiat Res 175:405–415. https://doi.org/10.1667/RR2461.1
Sokolov M, Panyutin IG, Neumann R (2006) Genome-wide gene expression changes in normal human fibroblasts in response to low-LET gamma-radiation and high-LET-like 125IUdR exposures. Radiat Prot Dosim 122:195–201
Amundson SA, Fornace AJ Jr (2001) Gene expression profiles for monitoring radiation exposure. Radiat Prot Dosim 97:11–16
Widel M, Lalik A, Krzywon A, Poleszczuk J, Fujarewicz K, Rzeszowska-Wolny J (2015) The different radiation response and radiation-induced bystander effects in colorectal carcinoma cells differing in p53 status. Mutat Res 778:61–70. https://doi.org/10.1016/j.mrfmmm.2015.06.003
Lyng FM, Howe OL, McClean B (2011) Reactive oxygen species-induced release of signalling factors in irradiated cells triggers membrane signalling and calcium influx in bystander cells. Int J Radiat Biol 87:683–695. https://doi.org/10.3109/09553002.2010.549533
Bahreyni-Toossi MT, Khademi S, Azimian H, Mohebbi S, Soleymanifard S (2017) Assessment of the dose-response relationship of radiation-induced bystander effect in two cell lines exposed to high doses of ionizing radiation (6 and 8 Gy). Cell J 19(3):434–442. https://doi.org/10.22074/cellj.2017.4343
Mohd Zainudin NH, Abdullah R, Rahman WN (2020) Bystander effect induced in breast cancer (MCF-7) and human osteoblast cell lines (hFOB 1.19) with HDR-brachytherapy. J Biomed Phys Eng 10:319–328. https://doi.org/10.31661/jbpe.v0i0.1135
Rezaei M, Kamran Samani R, Kazemi M, Shanei A, Hejazi SH (2021) Induction of a bystander effect after therapeutic ultrasound exposure in human melanoma: In-vitro assay. Int J Radiat Res 19(1):183–189. https://doi.org/10.29252/ijrr.19.1.183
Wakatsuki M, Magpayo N, Kawamura H, Held KD (2012) Differential bystander signaling between radioresistant chondrosarcoma cells and fibroblasts after X-ray, proton, iron ion and carbon ion exposures. Int J Radiat Oncol Biol Phys 84:e103–e108. https://doi.org/10.1016/j.ijrobp.2012.02.052
Anzenberg V, Chandiramani S, Coderre JA (2008) LET-dependent bystander effects caused by irradiation of human prostate carcinoma cells with X rays or alpha particles. Radiat Res 170:467–476. https://doi.org/10.1667/rr1312.1
Autsavapromporn N, Plante I, Liu C, Konishi T, Usami N, Funayama T, Azzam EI, Murakami T, Suzuki M (2015) Genetic changes in progeny of bystander human fibroblasts after microbeam irradiation with X-rays, protons or carbon ions: the relevance to cancer risk. Int J Radiat Biol 91:62–70. https://doi.org/10.3109/09553002.2014.950715
Yin X, Tian W, Wang L, Wang J, Zhang S, Cao J, Yang H (2015) Radiation quality-dependence of bystander effect in unirradiated fibroblasts is associated with TGF-b1-Smad2 pathway and miR-21 in irradiated keratinocytes. Sci Rep 5:11373
Chen B, Zhang P, Sun F, Li B, Chen Y, Pei S, Zhang Z, Manzoor R, Deng Y, Sun C, Sui L, Kong F, Ma H (2020) The mechanism of bystander effect induced by different irradiation in human neuroblastoma cells. Acta Astronaut 166:599–606. https://doi.org/10.1016/j.actaastro.2018.12.005
Fu J, Yuan D, Xiao L, Tu W, Dong C, Liu W, Shao C (2016) The crosstalk between α-irradiated Beas-2B cells and its bystander U937 cells through MAPK and NF-κB signaling pathways. Mutat Res 783:1–8. https://doi.org/10.1016/j.mrfmmm.2015.11.001
Yoshino H, Nawamaki M, Murakami K, Kashiwakura I (2019) Effects of irradiated cell conditioned medium on the response of human lung cancer cells to anticancer treatment in vitro. World Acad Sci J 1:92–97. https://doi.org/10.3892/wasj.2019.11
El-Din AAM, Abdelrazzak AB, Ahmed MT, El-Missiry MA (2017) Radiation induced bystander effects in the spleen of cranially-irradiated rats. Br J Radiol 90:20170278. https://doi.org/10.1259/bjr.20170278
Mendonca MS, Howard KL, Farrington DL, Desmond LA, Temples TM, Mayhugh BM, Pink JJ, Boothman DA (1999) Delayed apoptotic responses associated with radiation-induced neoplastic transformation of human hybrid cells. Cancer Res 59:3972–3979
Widel M, Przybyszewski WM, Cieslar-Pobuda A, Saenko YV, Rzeszowska-Wolny J (2012) Bystander normal human fibroblasts reduce damage response in radiation targeted cancer cells through intercellular ROS level modulation. Mutat Res 731:117–124
Desai S, Kobayashi A, Konishi T, Oikawa M, Pandey BN (2014) Damaging and protective bystander cross-talk between human lung cancer and normal cells after proton microbeam irradiation. Mutat Res 763–764:39–44
Wang X, Zhang J, Fu J, Wang J, Ye S, Liu W, Shao C (2015) Role of ROS-mediated autophagy in radiation-induced bystander effect of hepatoma cells. Int J Radiat Biol 91(5):452–458
Zhang J, Zhang Y, Mo F, Patel G, Butterworth K, Shao C, Prise KM (2021) The roles of HIF-1α in radiosensitivity and radiation-induced bystander effects under hypoxia. Front Cell Dev Biol. 9:637454
He M, Ye S, Ren R, Dong C, Xie Y, Yuan D, Shao C (2012) Cytochrome-c mediated a bystander response dependent on inducible nitric oxide synthase in irradiated hepatoma cells. Br J Cancer 106:889–895
He M, Dong C, Ren R, Yuan D, Xie Y, Pan Y, Shao C (2013) Radiation enhances the invasiveness of irradiated and non-irradiated bystander hepatoma cells through a VEGF-MMP2 pathway initiated by p53. Radiat Res 180(4):389–397
Xie Y, Tu W, Zhang J, He M, Ye S, Dong C, Shao C (2015) SirT1 knockdown potentiates radiation-induced bystander effect through promoting c-Myc activity and thus facilitating ROS accumulation. Mutat Res 772:23–29
Acknowledgements
The author SM would like to thank the Indian Council of Medical Research (ICMR) for providing the research Grant (3/1/3/JRF-2016/HRD). The authors also express gratitude to UGC-DAE Consortium for Scientific Research, Kolkata Centre, for providing all the necessary facilities.
Funding
Supported by the fellowship from the Indian Council of Medical Research [Award letter No: 3/1/3/JRF-2016/HRD].
Author information
Authors and Affiliations
Contributions
Conceptualization: SM, AC; Methodology: SM, AD; Formal analysis and investigation: SM; Writing—original draft preparation: SM; Writing—review and editing: SM; Funding acquisition: SM; Resources: SM, AC; Supervision: AC.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that there is no conflict of interest.
Ethical approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Mukherjee, S., Dutta, A. & Chakraborty, A. The cross-talk between Bax, Bcl2, caspases, and DNA damage in bystander HepG2 cells is regulated by γ-radiation dose and time of conditioned media transfer. Apoptosis 27, 184–205 (2022). https://doi.org/10.1007/s10495-022-01713-4
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10495-022-01713-4