
Abstract
Ingestion of aristolochic acids (AA) contained in herbal remedies results in a renal disease and, frequently, urothelial malignancy. The genotoxicity of AA in renal cells, including mutagenic DNA adduct formation, is well-documented. However, the mechanisms of AA-induced tubular atrophy and renal fibrosis are largely unknown. Epithelial cell death is a critical characteristic of these pathological conditions. To elucidate the mechanisms of AA-induced cytotoxicity, we explored AA-interacting proteins in tubular epithelial cells (TEC). We found that AA interacts with a mitochondrial enzyme glutamate dehydrogenase (GDH) and moderately inhibits its activity. We report that AA induces cell death in GDH-knockdown TEC preferentially via non-apoptotic means, whereas in GDH-positive cells, death was executed by both the non-apoptotic and apoptotic mechanisms. Apoptosis is an energy-reliant process and demands higher adenosine 5′-triphosphate (ATP) consumption than does the non-apoptotic cell death. We found that, after AAI treatment, the ATP depletion is more pronounced in GDH-knockdown cells. When we reduced ATP in TEC cells by inhibition of glycolysis and mitochondrial respiration, cell death mode switched from apoptosis and necrosis to necrosis only. In addition, in cells incubated at low glucose and no glutamine conditions, oxaloacetate and pyruvate reduced AAI-induced apoptosis our data suggest that AAI-GDH interactions in TEC are critical for the induction of apoptosis by direct inhibition of GDH activity. AA binding may also induce changes in GDH conformation and promote interactions with other molecules or impair signaling by GDH metabolic products, leading to apoptosis.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Aristolochic acid (AA), found in herbal extracts of various species of Aristolochia, is a natural mixture composed of aristolochic acid I (AAI) and aristolochic acid II (AAII). AAI and AAII have similar genotoxic and carcinogenic potential and, although both compounds are cytotoxic, AAI is solely responsible for the nephrotoxicity associated with AA nephropathy [1]. AA tubulotoxicity resulting in mitochondrial damage combined with progressive tubular atrophy is related to impaired regeneration and apoptosis of proximal tubular epithelial cells (PTEC), characterized by a nearly complete absence of dedifferentiated cellular transmembrane migration. During the chronic phase, interstitial mesenchymal cells accumulate in the areas of collagen deposits. It has also been demonstrated that AA tubulotoxicity leads to defective activation of antioxidative enzymes and mitochondrial damage [2].
The aim of the present study is to identify the mechanisms of AA cytotoxicity in renal epithelium. To achieve this goal, we first screened for the potential AA-interacting proteins in renal epithelium, resulting in the identification of glutamate dehydrogenase (GDH).
GDH is found in all living organisms. In eukaryotes, GDH is located within the inner mitochondrial matrix. This enzyme catalyzes the reversible oxidative deamination of l-glutamate to 2-oxoglutarate, using NADP + as the coenzyme to feed 2-oxoglutarate to the Krebs cycle. Although there is some debate about the directionality of this reaction, the high Michaelis constant (K m) for ammonium in the reductive amination reaction appears to prohibit the reverse reaction under physiological conditions in most organisms [3]. The renal GDH activity is second only to that in the liver [4]. In the kidney, GDH activity is highest in the proximal tubule segments and is regulated by acid–base loading in these parts of kidney only [5]. In rabbit renal tubules, activation of GDH results in increased glucose and glutamine synthesis [6].
GDH in animals, but not in plants and microorganisms, is allosterically regulated by many ligands. Because GDH is stimulated by ADP and l-Leucine and inhibited by GTP, NADH and ATP, the activity of the enzyme can be controlled by ATP availability as an energy source [7].
An understanding of the mechanisms of GDH regulation has become an important goal for investigators in view of the discovery that a mutation in this pathway leads to a genetic hypoglycemic disorder [8–10].
To search for modulators of GDH activity that may be used as therapeutic modalities, 30,000 compounds were analyzed and 21 compounds were selected for further testing [11]. Studies of the mechanisms of GDH regulation in the pancreas showed that GDH regulation is directly linked to insulin release, most likely via modulation of ATP synthesis [12]. In addition, in other tissues, oxidation of glutamate by GDH is linked to the increase in ATP synthesis as an energy source [13]. The role of GDH in carbohydrate and amino acid metabolism was shown using TSC −/− cells and glutamine as a carbon source. The reliance on glutamine in this cellular model is completely dependent on GDH but not on other glutamate metabolizing enzymes [14]. In renal epithelium, GDH plays a key role in ammoniagenesis. When isolated renal cortical tubules were incubated with 2-aminobicyclo [2,2,1] heptanes-2-carbocylic acid (BCH), a nonmetabolizable analog of leucine, the rate of ammoniagenesis from glutamine increased by 40% via the activation of GDH, but not of other enzymes participating in glutaminolysis [15]. These data indicate that GDH is important for the mitochondrial energy generation as well as for the carbohydrate and amino acid metabolism, in different biological systems.
Subsequent studies demonstrated that the AAI-induced inhibition of GDH activity in proximal tubular cells was accompanied by cell death, partly via apoptotic pathways. To better understand the role of AAI-GDH interactions in AA-induced cytotoxicity, we generated GDH knockdown cells. We found that AAI induced proportionally less apoptotic cell death in GDH knockdown cells as compared to HK-2 and GDH+ cells, while its total cytotoxicity in GDH+ and GDH knockdown cells was comparable. We propose that the observed switch of cell death mode may reflect a greater extent of ATP depletion in GDH knockdown as compared to GDH+ cells, possibly due to the more profound, GDH-independent inhibition of ATP synthesis in GDH− cells.
In addition, GDH-AAI interactions and a consequential change of molecular conformation might trigger apoptosis via communication with other mitochondrial proteins. Finally, the metabolic products of a GDH-catalyzed reaction may also serve as signal messengers for the regulation of apoptosis, mediating the effects of low GDH activity on cell death regulation.
Thus, apoptotic cell death is directly linked to GDH interactions with AAI, whereas non-apoptotic cell death may be initiated by the additional, still unidentified AAI activities.
Results
AAI binds specifically to GDH in mouse cortex and in human urothelial cells
To elucidate the mechanism of AA cytotoxicity in target cells, we performed a search for AAI interacting proteins in the two target tissues where AAI is metabolized and forms DNA adducts; namely mouse renal cortex and cultured human urothelial cells. We chemically modified AAI by attaching biotin to a carboxyl group of the molecule (bAAI) (Fig. 1a).To identify the proteins that form complexes with AAI in human cultured urothelial cells and in mouse cortex tissue, we incubated the cell lysates with bAAI. The generated bAAI-protein complexes were immobilized on streptavidin beads and, after washing, were eluted with boiling SDS buffer. The SDS-PAGE analysis of the eluate revealed polypeptides with apparent molecular mass of ~50 kDa (Fig. 1b). When non-biotinylated AAI was added to the protein extract together with bAAI, the yields of such polypeptides were reduced (Fig. 1b). These polypeptides were identified by mass spectrometry as IPI00114209 SWISS-PROT:P26443 Glud1 Glutamate dehydrogenase 1, mitochondrial (GDH). A total of 11 unique peptides were matched to the database entry, resulting in 21.1% coverage.

AAI interacts with GDH in mouse renal cortex and in human urothelial cells. a Chemical structure of AAI-biotin (bAAI). b Pull-down assay. bAAI was incubated with whole cell lysates of T24 cells (left gel) or mouse renal cortex (right gel) with or without non-labeled AAI, followed by the incubation with streptavidin-coated beads. Streptavidin-bound complexes were eluted by boiling in SDS sample buffer and loaded onto SDS-PAGE gel. c AA-GDH interactions. AAI or AAII were incubated overnight with purified bovine GDH in the presence of metal zinc powder. Portion of the reaction was boiled with SDS sample buffer and run on SDS-PAGE gel. Position of fluorescent nitroreduced form of AA bound to GDH was identified by UV transilluminator. Lane 1 GDH without AA, Lane 2 GDH incubated with AAI, GDH incubated with AAII
To evaluate the ability of AA to bind GDH in vitro, we incubated the purified bovine enzyme that was chemically activated by zinc dust, in the absence or presence of AAI or AAII. A fluorescent band (nitroreduced form of AA bound to GDH) was observed on SDS-PAGE gel under UV illumination in the lane in which the AAI + GDH reaction was loaded. A band formed by a fluorescent product with similar mobility was less intense in the lane loaded with AAII + GDH reaction, and fluorescence was undetectable when GDH was incubated with zinc dust alone. This band corresponded to a protein with apparent molecular mass slightly over 50 kDa (Fig. 1c).
AA modulates the GDH activity
AA inhibits the purified bovine GDH
We next studied the ability of AA to modulate GDH activity in vitro, using the purified bovine enzyme. The dose–dependent inhibition of GDH activity by AAI and AAII is shown in Fig. 2a. Using a modified Hill equation, we found an estimated IC50 for AAI of ~28 and ~38 μM for AAII.

GDH inhibition by AA. a The structural formula of aristolochic acids. For AAI R = OCH3, for AAII R = H. The dose-dependent inhibition of GDH activity by the AA. The lines represent the results of nonlinear least-squares fitting using a modified Hill equation. Average of 3 independent experiments. b GDH activity in the lysates of HK-2 cells pretreated for 18 h was assayed as described in the Materials and methods. Results of 4 independent experiments for AA1, AAII and 3 for EGCG with error bar indicating SD. *P < 0.05 compared to untreated cells
AAI inhibits GDH activity in proximal tubule cells
To explore the ability of AA to modulate GDH activity in proximal tubule cells, we treated HK-2 cells with two concentrations of AAI, with AAII or with the GDH inhibitor epigallocatechin-3-monogallate (EGCG). Using the lysates of pretreated HK-2 cells, we found that the inhibitory effect of GDH was more pronounced in the cells that were pretreated with AAI compared to AAII or EGCG (Fig. 2b).
AA causes cell death in parental HK-2 and GDH knockdown cells
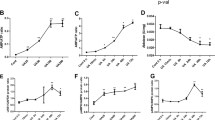
AAI induced cell death in HK-2 cells in a dose-dependent manner (Fig. 3a). We also found that AAI was more toxic in these cells than is AAII or the GDH inhibitor, EGCG. Another known GDH inhibitor, GW 5074, shows toxicity comparable to that of AAI in these cells (Fig. 3b). To study the mechanisms of AA-induced cell death and the role of GDH in this process, we established a GDH knockdown cell line, by using a GDH specific shRNA. The combined stable clones, transduced with shRNA for GDH, were designated as GDH− cells. When GDH was transiently reintroduced into GDH− cells (designated as GDH−+) they showed resistance to AAI in cytotoxicity assays, comparable to that of GDH+ and GDH− cells (Fig. 3c, d). During the incubation, the GDH+, GDH− and GDH+− cells exhibited comparable reduction of viable cell counts (Fig. 3d). For the experiments where precise quantitation of viable and apoptotic cells was critical, under numerous experimental conditions, we used the ApoTox-Glo™ Triplex assay. This system combines three Promega assay chemistries to assess viability, cytotoxicity and caspase activation events within a single assay well. The live-cell protease activity is restricted to intact viable cells and is measured using a fluorogenic, cell-permeant, peptide substrate (glycylphenylalanyl-aminofluorocoumarin; GF-AFC) [16]. We measured the numbers of viable cells (% of non-treated control) considering the rest of cells in population as non-viable. The non-viable cell subpopulation contains apoptotic cells as well as those that died via non-apoptotic mechanisms. However, this approach cannot further distinguish between the necrotic cells and those that died via a mechanism that is neither necrotic nor apoptotic.

Cell death induced in renal epithelial cells by AA. a Dose-dependent cell viability was assayed with ApoTox-Glo™ Triplex assay. HK-2 cells were exposed to AAI for 18 h. All data are representative of three independent experiments. b Cell death (ApoTox-Glo™ Triplex assay bis-AAF R110). HK-2 cells were exposed to AA and GDH inhibitors for 18 h. All data are representative of three independent experiments. c GDH protein amounts were analyzed by means of immunoblotting. HK-2 cells were infected with shRNA constructs targeting GDH (GDH−). GFP is HK-2 cells infected with lentivirus containing shRNA targeting GFP (negative control). Stable clone were selected and combined. GDH− cells transiently transfected with plasmid carrying full length cDNA for GDH were designated as GDH−+. Actin is used as loading control (immunoblotting with anti β actin antibodies). d HK-2 or shRNA GFP (GDH+) and shRNA GDH (GDH−) cells were incubated in presence of 40 μM AAI for indicated time. Cell viability was measured with ApoTox-Glo™ Triplex assay (viability assay GF-AFC). All data are representative of three independent experiments
To better distinguish between the viable, necrotic and apoptotic cells after AAI treatment, we analyzed the Hoechst 33342 and PI-stained cells by fluorescent microscopy. The morphological criteria defining the cell death type upon staining were as follows: the nuclei of viable cells stain blue (the cytoplasm remains invisible); apoptotic cells can be recognized by condensed chromatin, which usually stained more intensely blue; a similar chromatin condensation is observed at the late stage of apoptosis, but the color shifts to pink due to PI intrusion through leaky membranes; pink colored nuclei without chromatin condensation are typical of necrotic cells [17]. Using this approach, we found that most of the non-apoptotic dead cells were necrotic. However, we chose to use the term “non-apoptotic” (instead of “necrotic”) for the further interpretation of the data obtained in our cell viability assays with GF-AFC.
The proportion of apoptotic cells present in the population of dead cells was determined by measuring the caspase 3/7 (Fig. 4a) and caspase 9 (Fig. 4b) activities. In addition, apoptotic cells were identified by their distinct nuclear morphology using Hoechst/PI staining (Fig 4c), as well as by cytochrome c release assays (Fig. 4d).

Apoptosis induced in renal epithelial cells. a GDH+, GDH− and GDH−+ cells were incubated with AAI (40 μM) for indicated time. Apoptosis was assayed by measuring caspase 3/7 activity. All data are representative of at least three independent experiments. b GDH+ and GDH− cells were incubated with AAI (40 μM) for indicated time. Apoptosis was assayed by measuring caspase 9 activities. All data are representative of two independent experiments. c Left panel non treated GDH+ cells, in the middle GDH+ cells incubated with AAI (40 μM) for 12 h: right panel GDH-cells incubated with AAI (40 μM) for 12 h. Cells were stained with Hoechst 33342, and analyzed by fluorescence microscopy. White arrows indicate apoptotic cells. d Cytochrome c release from mitochondria. HK-2 cells were incubated with or without AAI (40 μM) for 18 h. Cytochrome C was measured after cell fractionation in cytoplasmic and mitochondrial fraction by cytochrome C ELISA. Representative results of 2 independent experiments. e GDH+ and GDH− cells were incubated with AAI, EGCG for 18 h or with STS for 4 h. Apoptosis was assayed by measuring caspase 3/7 activity. The average data of 2 independent experiments. Error bars indicate SD
When GDH+ cells were treated with AAI, the number of apoptotic cells increased during incubation. In GDH knockdown cells, we observed an increase in the total number of dead cells, while the proportion of apoptotic cell in the pool of dead cells did not changed substantially (Figs. 3d, 4a, b). In GDH+− cells in the presence of AAI, the number of apoptotic cells increased during incubation (Fig. 4a). Similar effects were observed with another GDH inhibitor, EGCG, albeit at different drug concentrations; whereas staurosporin (STS), a compound that has no GDH-specific activity, efficiently induced apoptosis in both GDH+ and GDH− cells (Fig. 4e).
ATP depletion is a potential cause of AA-induced cell death
Unlike necrosis, apoptosis is an energy-dependent process. Since the HK-2 GDH+ cells treated with AAI died partly via apoptosis, but AAI induced substantially less apoptosis in GDH− cells, we compared the dynamics of ATP reduction that was induced by 40 μM of AAI in GDH+ and GDH− cells. We found that ATP was substantially more depleted in GDH− cells than in GDH+ cells under the same experimental conditions. The ATP concentration in GDH+ cells after incubation with 40 μM AAI for 18 h was ~75% of that observed in non-treated cells. However, in GDH− cells treated at the same conditions, the ATP levels were ~45% of those in non-treated cells. These effects were more pronounced with longer incubation periods (Fig. 5).

ATP deficiency induced in GDH+, GDH− cells by AA1 (40 μM) treatment for indicated times. Experiments were carried out in triplicate. *P < 0.01 Values in GDH− set compared to related bars in GDH+ set
Graded ATP depletion in HK2 cells causes necrotic or apoptotic cell death
To test the idea that the differences in ATP content in AAI-treated GDH+ and GDH− cells are responsible for the choice of the cell death mode, we performed ATP depletion in HK-2 cells, by using various combinations of a glycolysis inhibitor DOG; dextrose; and/or inhibition of mitochondrial respiration with FCCP. We found that the ATP levels in HK-2 cells were indeed gradually reduced from stage 1 to stage 6: 75% under mild conditions (stage 1: 2 mM DOG + 6 mM dextrose) to 8% at severe conditions (stage 6: 2 μM FCCP) (Fig. 6a).

Chemically-modulated ATP depletion in HK-2 cells induces necrosis and apoptosis. a graded ATP depletion by combination of glycolysis or mitochondrial respiration inhibitors combined with limited dextrose conditions. Cells were treated as indicated for 18 h. Data are representative of three independent experiments. Error bars-SD. b Induction of cell death by ATP depletion. Cells were treated as indicated for 18 h and were assayed by H33342/PI staining and fluorescent microscopy for viable, dead and apoptotic cells. Cells were manually counted. At least 80 cells were counted for each sample. The average data of 2 independent experiments
We calculated the numbers of viable, necrotic and apoptotic cells at these 6 stages of ATP depletion. In these experiments, we used the H-33342/PI staining and fluorescent microscopy to analyze the cell death mode. The criteria for distinguish the viable, necrotic and apoptotic cells are described in the section “AA causes cell death in parental HK-2 and GDH knockdown cells”. At the first four stages of ATP depletion (stage 1: 6 mM dextrose, stage 2: 2 mM DOG + 6 mM dextrose, stage 3: 2 μM FCCP + 6 mM dextrose, stage 4: 2 mM DOG), we observed graded increases of dead cells that were mostly apoptotic. When ATP level reached 50% (stage 5: 2 μM FCCP + 4 mM dextrose), the numbers of dead cells further increased, due to both necrosis and apoptosis. At the lowest ATP level (stage 6: 2 μM FCCP), we found that the dead cells were primarily necrotic (Fig. 6b). In parallel, we performed quantitation of viable and apoptotic cells using ApoToxGlo Triplex assay as described above for viability studies. The amounts of necrotic cells determined using Hoechst/PI staining were comparable to the amounts of dead cells as measured by ApoToxGlo Triplex assay.
Apoptosis caused by AAI is reduced by the Krebs cycle substrates
Since ATP is generated via glycolysis and oxidative phosphorylation, we reasoned that the ATP production via glutaminolysis could be regulated, at least in part, by AAI modulation of GDH activity. GDH and alanine and/or aspartate amino transferases are responsible for the conversion of glutamate into 2-oxoglutarate, a Krebs cycle intermediate. Inhibition of GDH activity by AAI may cause a deficiency of 2-oxoglutarate, further leading to ATP depletion and, subsequently, cell death. In addition to its role as a source of NADH for oxidative phosphorylation, the Krebs cycle is also an important participant in many other metabolic pathways, and deficiencies in the Krebs cycle intermediates could lead to apoptosis, even when substantial amounts of ATP are provided by glycolysis [18].
Therefore, we examined the influence of Krebs cycle intermediates on AAI-induced apoptosis in HK-2 cells at Gln− and Gln+ conditions. The Gln− conditions were tested because they render intracellular glutamate the only source for α ketoglutarate synthesis via GDH. The absence of Gln in culture media did not induce apoptosis by itself (Fig. 7) but diminished cell viability (data not shown). We added oxaloacetate (OXA), a membrane-permeable Krebs cycle intermediate; dimethyl α ketoglutarate (DMαKG), a membrane permeable derivative of α ketoglutarate; or pyruvate, which can enter the cycle via conversion into acetyl-CoA or OXA in incubation media of AAI-pretreated cells. We found that, for cells incubated in media containing dextrose (d-glucose) and glutamine, apoptosis was not prevented with all assayed intermediates. For cells incubated in media containing low glucose and no glutamine OXA and pyruvate reduced AAI-induced apoptosis (Fig. 7).

Krebs cycle intermediates reduced apoptosis induced by AAI in GDH+ cells incubated with and without glutamine. Cells were treated with AAI (40 μM) with or without OXA (2 mM), DM αKG (7 mM) or pyruvate (2 mM).Apoptosis was assayed 18 h post treatment via measuring caspase 3/7 activities.SD is the average of 3 independent experiments. *P < 0.05. Values for intermediates and AAI added conditions compared with AAI treatment along
Discussion
In this study, we show that AAI specifically interacts with, and moderately inhibits GDH. This has been demonstrated with the purified enzyme as well as in the lysates from HK-2 cultured renal epithelial cells. AAI, more efficiently than EGCG, another GDH inhibitor, reduces GDH activity in renal epithelial cells, although EGCG is a more potent GDH inhibitor (IC50 500 nM) of purified GDH [10]. AAI was also more toxic to cultured renal epithelial cells than was EGCG. Thus, the specific intracellular environment of renal epithelial cells might be an important factor in AAI processing and its related activity.
When compared with AAII, AAI is a stronger inhibitor of GDH and is more toxic to renal epithelial cells. As these two compounds differ in the structure of the groups at position R (Fig. 2a), this group may be important in the development of GDH inhibition and renal cytotoxicity. Indeed, in the structure of nitrophenanthrene carboxylic acid compounds the “nitro” (–NO2) group and “methoxy” (–OCH3) group in the R position are critical determinants of maximum toxicity as observed for AAI, which is an 8-methoxy-6-nitro-phenanthro-(3,4-d)-1,3-dioxolo-5-carboxylic acid. Any modification of the structure of AAI, namely the addition, deletion, substitution, or replacement of these side chains drastically reduces toxicity. In AAII, the removal of the “methoxy” group from its position reduces the toxicity of AAII, a compound that differs from AAI by one “methoxy” group [17]. Therefore, specificity of AA and its activity are dependent on the chemical structure and conformation of particular AA specie as well as on the intracellular environment in specific tissue.
AA induced cell death
Epithelial cell death is an important component of the pathophysiology of tubular atrophy and, possibly, renal interstitial fibrosis. Therefore, we studied cell death induced by AA in tubular epithelial cells. To further elucidate the role of GDH-AA interactions, we generated GDH knockdown HK-2 cells. Survival rates of these cells without treatment were not impaired. Untreated GDH− cells did not exhibit elevated levels of apoptosis, although we did observe apoptosis when GDH activity was inhibited by AAI in GDH+ cells. This disparity might be explained by the de novo activation of energy metabolic pathways in GDH− cells that are alternative to the GDH-dependent pathways in GDH+ cells. Such activation may compensate for the ATP insufficiency caused by the GDH deficiency. When incubated with AAI, GDH+ and GDH− cells show similar kinetics of cell demise, whereas the number of apoptotic cells in the pull of all dead cells after this treatment increased in GDH+ but not in GDH− cells.
Apoptosis induced by AA
Apoptosis is a form of programmed cell death that is executed by a family of proteases called caspases that can be activated either by cell surface death receptors (extrinsic pathway) or by perturbation of mitochondrial membrane (the intrinsic pathway) [19]. The intrinsic (mitochondrial) pathway is characterized by the release of cytochrome c from mitochondria in cytoplasm and by the activation of caspase 9. As we observed the both phenomena as well as mitochondrial membrane permeability change in cells treated with AA, we concluded that AA induced apoptosis, at least partly, by intrinsic pathway.
Cell death and, in particular, apoptosis, is often a p53-dependent process [20]. Previously, it was shown that AAI induced massive apoptotic and necrotic tubular epithelial cell death and acute renal failure in wild-type mice and in cultured cells, whereas p53 deficiency (knockout mice) and pharmacologic inhibition of p53 by specific inhibitor pifithrin-α in vitro attenuates this injury [21, 22]. We observed phosphorylation of p53 on serine-15 in HK-2 cells in response to AAI treatment at the conditions (40 μM, 18 h), that induced cell death partly via apoptosis in these cells. Therefore, apoptosis, induced in renal epithelial cells by AAI is p53 dependent.
In GDH+ cells, AA likely induces non-apoptotic and apoptotic cell death by two different mechanisms. Non-apoptotic death in these cells is initiated by a yet unidentified mechanism; apoptotic cell death may be related to GDH inhibition by AA.
In addition to the proposed mechanism for AAI induction of apoptotic cell death in HK-2 cells, AAI interaction with GDH might be linked to AA cytotoxicity in renal tubular epithelial cells by other means. For example, several metabolites whose synthesis is regulated by GDH, namely, glutamine, glutamate and 2-oxoglutarate, are good candidates to be intracellular (mitochondrial) messengers. It is possible that altered synthesis of these molecules could activate other mitochondrial triggers for apoptosis induction. Thus, it is known that mitochondrial glutamate acts as a messenger in glucose-induced insulin secretion [23]. In GDH knockdown cells, the presence of AAI results in more dramatic and nonspecific changes, whereas in GDH+ cells, a less significant difference of metabolite production might be a signal for apoptosis induction. Alternatively, changes in the conformation of GDH after interaction with AA might initiate communication with other molecules. For instance, it was shown that during execution of TNF-induced necrosis, the protein kinase receptor-interacting protein 3 (RIP3) became associated with GDH [24].
Different ATP depletion induced by AAI in renal epithelial cells as a cause of specific cell death mode
It was shown for several cell systems that graduated ATP depletion changed the mode of cell demise [25–27]. Moreover, selective and graded repletion of the ATP pool with glucose restores the ability of the cells undergo apoptosis [26]. We observed the change of cell death mode when ATP depletion was performed in HK-2 cells using combination of glycolysis, mitochondrial inhibitors and different glucose amount in culture media. The complete apoptotic process involves energy-requiring steps, such as caspase activation, enzymatic hydrolysis, bleb and apoptotic body formation. A deficiency of ATP at these stages would prevent downstream steps of the apoptotic pathway. In addition, low intracellular levels of free ATP might permit apoptotic stimuli to induce necrosis [28].
As alteration of ATP synthesis is the most direct consequence of GDH inhibition, and ATP was depleted in GDH+ and GDH− cells treated with AAI in different way, we propose that this difference might be a cause of the observed changes in the mode of cell death in these 2 cell types.
The deficiency in Krebs cycle as a consequence of AAI action resulted in ATP insufficiency and cell death
To link GDH inhibition by AAI to glutamine metabolism via GDH we compared AAI action at Gln free and Gln+ conditions. The absence of glutamine in culture media did not induce apoptosis but impaired cell survival. OAA and pyruvate were partly able to reduce AAI- induced apoptosis at these conditions. With limited glutamine source GDH and aminotransferases may metabolize only intracellular glutamate to provide α ketoglutarate to Krebs cycle. At these conditions activity of GDH could be more critical for ATP production and its inhibition by AAI resulted in stronger apoptosis than in the presence of Gln. The compensation of this deficiency by external OXO and pyruvate may provide more ATP production and partly prevent apoptosis induced by the ATP deficiency. In tested cellular system OXO and pyruvate prevent apoptosis more efficiently than α ketoglutarate, possibly because of the weak permeability of mitochondrial membranes for DM αKG or insufficient metabolic transformation of DM αKG in these cells. Therefore, AAI cytotoxicity in tubular epithelial cells resulted in Krebs cycle deficiency, may be partly prevented by Krebs cycle intermediates.
Conclusions
In light of the fact that mechanisms of AA-induced tubular atrophy and interstitial fibrosis are largely unknown and cell death is a critical component of these pathologic conditions, our results point to the important fact that AA-GDH interaction may play a critical role in apoptosis induction by AA in renal epithelial cells. Specifically, inhibition of GDH activity might result in a decrease in ATP production, which is necessary to trigger apoptosis, but still maintains an ATP level sufficient to execute apoptosis.
Therefore, in tubular renal epithelial cells AAI interacts with GDH, inhibits its activity and induces cell death via apoptotic and non-apoptotic mechanisms. The induced apoptosis is directly linked to AAI-GDH interactions. Non-apoptotic cell death (mostly necrosis) is induced by unidentified yet mechanisms. In addition to the direct enzymatic inhibition of GDH by AAI, its conformational change may initiate communications with other mitochondrial enzymes. To our knowledge, this is the first indication of GDH involvement in the regulation of apoptosis and of the role of GDH in AAI-induced renal tubulotoxicity. Further exploration of the consequences of AA-GDH interactions will shed light on the mechanism of AA cytotoxicity induced by activation of GDH in renal epithelium.
Materials and methods
Cell lines and reagents
Immortalized human proximal tubule epithelial HK-2 and urothelial T24 cells (American Type Culture Collection) were maintained in RPMI-1640 supplemented with 4.5 g/l d-glucose, 2 mM l-glutamine, 10% fetal bovine serum, and mixture of penicillin and streptomycin (100 mg/ml each). Cells were used between passages 10–29. Cells were trypsinized and seeded onto 6-well plates, glass coverslips on 12-well plates or 96-well plates, depending on experimental conditions. Cells were grown to full confluency before treatment to represent the in vivo situation of differentiated epithelial monolayers. Anti-actin and anti-GDH monoclonal antibodies and all other reagents were obtained from Sigma-Aldrich (St. Louis, MO).
Biotinylation of AAI
To identify AA binding molecules, we synthesized a biotinylated derivative of AAI using a carboxyl group for biotin attachment via spacer arm. The synthesis was performed using EZ-Link Amine-PEG2-biotin reagent (Pierce) based on the procedure described by Tsuchihara et al. [29] for the synthesis of fluorescent derivative of AAI with some modifications. Briefly: AA was reacted with tert-butyl bromo acetate in the presence of 18 crown ether/K2CO3 in a mixture of acetone/acetonitrile for 1 h at reflux temperature to give the corresponding tert-butyl ester which was purified by silica gel column chromatography. The tert-butyl ester was deprotected using trifluoroacetic acid to give the corresponding acid. The acid was then reacted with biotin protected amine derivative in the presence of EDC.HCl/DIPEA to give the crude biotinylated AA derivative which was purified by HPLC.
Affinity purification of AAI binders from mouse kidney and human cultured cells
Affinity purification was performed using streptavidin-agarose or streptavidin magnetic beads (Pierce). Briefly: 100 μl of bAAI in TBS was first preincubated overnight at 4°C with streptavidin-agarose as described by manufacturer (Pierce). Unbound bAAI was removed by several washes with TBS. Cultured T24 cells were disrupted in a lysis buffer (50 mM Tris–HCl, pH 7.5., 150 mM NaCl, 1% Nonidet P-40, 0.5% sodium deoxycholate and protease inhibitor tablet (Roche); mouse kidney cortex was homogenized in lysis buffer. Portion of lysates were preincubated with streptavidin beads to remove unspecific binders. Precleaned lysates were incubated with bAAI loaded beads overnight at 4°C with different salt content (0.1–3 M). Biotin-free AAI was added to some lysate portions to compete for prey protein binding to immobilized bAAI. After the binding, beads were washed several times with lysis buffer and bound molecules were eluted by boiling for 5 min in SDS sample buffer. Eluates were run on SDS PAGE gels. Gels were stained with Silver Colloidal stain (Pierce).
AA binding to GDH in vitro
Purified bovine GDH (10 μM) was treated in vitro with 0.12 and 1.2 mM AAI or AAII in 250 μl 50 mM potassium phosphate buffer, pH 5.8, in the presence of 5 mg zinc dust for chemical activation [30]. The reaction mixtures were incubated overnight on a rocking platform at room temperature. The aliquot of reaction mixture was mixed with equal amount of 2X SDS sample buffer, boiled for 5 min sample buffer and run on an SDS PAGE gel. The position of reduced AA (fluorescent) bound to GDH was detected under UV illumination.
Kinetic analysis
Kinetic analysis was performed basically as described [11]. The IC50 for AA1 and AA2 were measured in the oxidative deamination directions in 0.1 M sodium phosphate buffer, pH 8.0, using 50 mM sodium glutamate and 0.2 mM NAD+. The curves were fitted to a modified Hill equation:
where V i is the maximum inhibition caused by the drug (%) and IC50 is the concentration of drug that caused half-maximal inhibition of the reaction, h is the Hill coefficient [11]. Data was fitted using the nonlinear regression procedure in the program Prism 4 (GraphPad Software, Inc. La Jolla, CA).
Enzymatic activity in cell lysates
GDH activity in cell lysates was assayed in the direction of deamination. Cells (1 × 106) were washed twice with ice–cold PBS and lysed with 500 μl of lysis buffer (20 mM Tris–HCl pH 7.5, 150 mm NaCl, 1% Triton X-100, 2 mM EDTA, 1 mM Na3VO4, and protease inhibitors) for 20 min on ice. Lysates were precleaned by centrifugation (12.000×g for 15 min at 4°C). Amount of lysate containing 0.5 mg of protein was incubated in reaction buffer with 20 mm glutamate and 5 mM NAD+. Activity was measured by the change in NADH fluorescence at 352/464 nm in a microplate reader (Molecular Devices).
Cell death, viability, apoptosis assays
ApoTox-Glo Triplex assay and fluorescent measurement of caspase 3, 7 and 9 activity
The ApoTox-Glo™ Triplex Assay combines three Promega assays to assess viability, cytotoxicity and caspase activation events within a single assay well. The first part of the assay simultaneously measures two proteases activities: one is a marker of cell viability and the other is a marker of cytotoxicity. The live-cell protease activity is restricted to intact viable cells and is measured using a fluorogenic, cell-permeant, peptide substrate (GF-AFC). A second, fluorogenic cell-impermeant peptide substrate (bis-alanylalanyl-phenylalanyl-rhodamine 110; bis-AAF-R110) is used to measure dead-cell protease activity, which is released from cells that have lost membrane integrity. Because bis-AAF-R110 is not cell-permeant, essentially no signal from this substrate is generated by intact viable cells. The second part of the assay uses the Caspase-Glo® Assay Technology by providing a luminogenic caspase-3/7 substrate, which contains the tetrapeptide sequence DEVD, in a reagent optimized for caspase activity, luciferase activity and cell lysis (Promega Corp., Madison, WI). Alternatively, caspase 3/7 or 9 activity was measured with fluorigenic substrates Ac-DEVD AFC and Ac-LEHD AFC, respectively, following the manufacturer’s protocol (BioVision, CA).
Hoechst 33342 and propidium iodide (PI) staining
To analyze mode of cell death cells we stained cells with membrane permeable Hoechst 33342 (excitation wavelength, 348 nm; emission wavelength, 479 nm) along or together with DNA dye PI (excitation wavelength, 500 nm; emission wavelength, 640 nm) generally as described [17]. Briefly: cells were grown on 18 mm cover slips. Cells were washed with PBS and H-33342 and PI (1 μg/ml) in PBS were added. After incubation for 30 min at 37°C cells were fixed with 3.7% paraformaldehyde solution in PBS. Cover slips were mounted on microscopic slides and samples were examined with Nikon fluorescent microscope. The Hoechst stain the nuclei of all cells, cells with condensed chromatin and nuclei fragmentation were scored as apoptotic. PI can penetrate cells with the lost membrane integrity, characteristic of necrotic and late apoptotic cells. Necrosis is characterized by nuclear PI uptake without chromatin condensation.
Cellular fractionation and western blot analysis
Isolation of the mitochondrial and cytosolic fractions was performed as previously described [31]. Briefly, 108 cells were harvested and washed twice in PBS before washing in lysis buffer (250 mM sucrose, 20 mM HEPES/KOH, pH 7.5, 50 mM KCl, 2.5 mM MgCl2, 1 mM DTT, and a tablet of complete protease inhibitor mixture; (Roche, Indianapolis, IN). After centrifugation, the pellet was resuspended in an equal volume of lysis buffer and placed on ice for 30 min. The cells were then homogenized with a Potter–Elvehjem Teflon-coated pestle (50–100 strokes), until 50% of them became permeable to trypan blue and immediately spun down twice for 5 min at 300×g to remove nonlysed cells and nuclei. A final centrifugation at 15,000×g for 15 min separated cytosol (supernatant) from the heavy membrane pellet, containing mitochondria [32]. The pellet was resuspended in a volume equal to that of the supernatant before protein quantification (Bradford assay) and ELISA. Equivalent aliquots of cytosol and mitochondrial extracts were loaded in ELISA plate wells precoated with cytochrome C antibodies (Quantikine M., R&D Systems). Alternatively, a portion of total cell lysates was loaded on SDS-PAGE and processed for Western blot.
ATP measurement
ATP was extracted by adding 1 ml of boiling water to the cell pellet as described [33]. The extract was vortexed and cleared for 5 min at 12,000 g at 4°C. 700 μl of the cleared supernatant were taken to measure ATP by the Luciferin-Luciferase kit (Sigma-Aldrich) following the manufacturer’s instructions. The pellet was resuspended in the remaining 300 μl of supernatant and used to determine protein concentration. The concentration of protein was also determined in the supernatant used to determine ATP concentration to correct for any protein left after centrifugation of the extract. The amount of protein in both fractions was combined to obtain the total protein amount in the extract [34].
ATP depletion of HK-2 cells using inhibitors of glycolysis and mitochondrial respiration
ATP deficiency conditions were created in HK2 cells as described, with some variations [24]. HK2 cells were incubated in dextrose-free DMEM with 2 mM of glutamine. ATP depletion of varying degrees was induced by adding cyanide-p-trifluoromethoxyphenylhydrazone (FCCP) (2 μM, a mitochondrial inhibitor) or 2-deoxyglucose (DOG) (5 mM, inhibitor of glycolysis) with and without dextrose. Six different metabolic conditions were examined: #1. (FCCP2 μM) without dextrose, #2. FCCP + 2 mM dextrose, #3. FCCP + 6 mM dextrose, #4. DOG, #5. DOG + 6 mM dextrose, #6. 6 mM dextrose
shRNA knockdown of GDH and transfection of GDH− cells with GDH
Cells were seeded at a density 50–70% in 12 well plates and were infected with lentivirus coding shRNA specific for GLUD1 (GDH) sc-90633-v or with virus coding GFP (negative control) (Santa Cruz Biotechnology). After transduction, stable cell lines expressing the shRNA were isolated via selection with puromycin. To reestablish GDH in GDH knockdown cells, the pCMV6-XL5 plasmid containing full-size GDH cDNA coding human Glud1 (SC116830) was transfected into GDH− cells using Lipofectamine 2000 tranfection reagent. Transiently transfected cells were assayed for cell viability and apoptosis with and without AAI treatment.
Statistical analysis
All values in the figures are expressed as the mean ± SD. The number of performed experiments is listed in the figure legends. Differences were analyzed statistically using the t-test. Analysis was accomplished with Prism 4 (GraphPad Software, Inc. La Jolla, CA).
Abbreviations
- NADP+:
-
Nicotinamide adenine dinucleotide phosphate
- ATP:
-
Adenosine 5′-triphosphate
- GTP:
-
Guanosine 5′-Triphosphate
- ADP:
-
Adenosine diphosphate
- SDS:
-
Sodium dodecyl sulfate
- PAGE:
-
Polyacrylamide gel electrophoresis
- shRNA:
-
Short hairpin ribonucleic acid
- OXPHOS:
-
Oxidative phosphorylation
- NAD:
-
Nicotinamide adenine dinucleotide
- EDTA:
-
Ethylenediaminetetraacetic acid
- DTT:
-
Dithiothreitol
- HEPES:
-
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- EGCG:
-
Epigallocatechin-3-monogallate
- Gln:
-
Glycine
- DOG:
-
2-deoxyglucose
- DM-αKG:
-
Dimethyl α ketoglutarate
- FCCP:
-
Cyanide-p-trifluoromethoxyphenylhydrazone
References
Shibutani S, Dong H, Suzuki N, Ueda S, Miller F, Grollman AP (2007) Selective toxicity of aristolochic acids I and II. Drug Metab Dispos 35:1217–1222
Pozdzik AA, Salmon IJ, Debelle FD et al (2008) Aristolochic acid induces proximal tubule apoptosis and epithelial to mesenchymal transformation. Kidney Int 73:595–607
Smith TJ, Stanley CA (2008) Untangling the glutamate dehydrogenase allosteric nightmare. Trends Biochem Sci 33:557–564
Treberg JR, Brosnan ME, Watford M, Brosnan JT (2010) On the reversibility of glutamate dehydrogenase and the source of hyperammonemia in the hyperinsulinism/hyperammonemia syndrome. Adv Enzyme Regul 50:34–43
Wright PA, Knepper MA (1990) Glutamate dehydrogenase activities in microdissected rat nephron segments: effects of acid-base loading. Am J Physiol 259:F53–F59
Winiarska K, Bozko P, Lietz T, Bryla J (1998) Importance of glutamate dehydrogenase stimulation for glucose and glutamine synthesis in rabbit renal tubules incubated with various amino acids. Acta Biochim Pol 45:825–831
Plaitakis A, Zaganas I (2001) Regulation of human glutamate dehydrogenases: implications for glutamate, ammonia and energy metabolism in brain. J Neurosci Res 66:899–908
Stanley CA (2009) Regulation of glutamate metabolism and insulin secretion by glutamate dehydrogenase in hypoglycemic children. Am J Clin Nutr 90:862S–866S
Li C, Matter A, Kelly A et al (2006) Effects of a GTP-insensitive mutation of glutamate dehydrogenase on insulin secretion in transgenic mice. J Biol Chem 281:15064–15072
Li C, Allen A, Kwagh J et al (2006) Green tea polyphenols modulate insulin secretion by inhibiting glutamate dehydrogenase. J Biol Chem 281:10214–10221
Li M, Allen A, Smith TJ (2007) High throughput screening reveals several new classes of glutamate dehydrogenase inhibitors. Biochemistry 46:15089–15102
Li M, Smith CJ, Walker MT, Smith TJ (2009) Novel inhibitors complexed with glutamate dehydrogenase: allosteric regulation by control of protein dynamics. J Biol Chem 284:22988–23000
Smith JE, Hall PV, Galvin MR, Jones AR, Campbell RL (1979) Effects of glycine administration on canine experimental spinal spasticity and the levels of glycine, glutamate, and aspartate in the lumbar spinal cord. Neurosurgery 4:152–156
Choo AY, Kim SG, Vander Heiden MG et al (2010) Glucose addiction of TSC null cells is caused by failed mTORC1-dependent balancing of metabolic demand with supply. Mol Cell 38:487–499
Treberg JR, Clow KA, Greene KA, Brosnan ME, Brosnan JT (2010) Systemic activation of glutamate dehydrogenase increases renal ammoniagenesis: implications for the hyperinsulinism/hyperammonemia syndrome. Am J Physiol Endocrinol Metab 298:E1219–E1225
Niles AL, Moravec RA, Eric Hesselberth P, Scurria MA, Daily WJ, Riss TL (2007) A homogeneous assay to measure live and dead cells in the same sample by detecting different protease markers. Anal Biochem 366:197–206
Grusch M, Polgar D, Gfatter S et al (2002) Maintenance of ATP favours apoptosis over necrosis triggered by benzamide riboside. Cell Death Differ 9:169–178
Albayrak T, Scherhammer V, Schoenfeld N et al (2003) The tumor suppressor cybL, a component of the respiratory chain, mediates apoptosis induction. Mol Biol Cell 14:3082–3096
Lewis JS, Meeke K, Osipo C et al (2005) Intrinsic mechanism of estradiol-induced apoptosis in breast cancer cells resistant to estrogen deprivation. J Natl Cancer Inst 97:1746–1759
Amaral JD, Xavier JM, Steer CJ, Rodrigues CM (2010) The role of p53 in apoptosis. Discov Med 9:145–152
Zhou L, Fu P, Huang XR, Liu F, Lai KN, Lan HY (2010) Activation of p53 promotes renal injury in acute aristolochic acid nephropathy. J Am Soc Nephrol 21:31–41
Zhou L, Fu P, Huang XR et al (2010) Mechanism of chronic aristolochic acid nephropathy: role of Smad3. Am J Physiol Renal Physiol 298:F1006–F1017
Maechler P, Wollheim CB (1999) Mitochondrial glutamate acts as a messenger in glucose-induced insulin exocytosis. Nature 402:685–689
Zhang DW, Shao J, Lin J et al (2009) RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science 325:332–336
Leist M, Single B, Castoldi AF, Kuhnle S, Nicotera P (1997) Intracellular adenosine triphosphate (ATP) concentration: a switch in the decision between apoptosis and necrosis. J Exp Med 185:1481–1486
Lieberthal W, Menza SA, Levine JS (1998) Graded ATP depletion can cause necrosis or apoptosis of cultured mouse proximal tubular cells. Am J Physiol 274:F315–F327
Nicotera P, Leist M, Ferrando-May E (1999) Apoptosis and necrosis: different execution of the same death. Biochem Soc Symp 66:69–73
Miyoshi N, Watanabe E, Osawa T et al (2008) ATP depletion alters the mode of cell death induced by benzyl isothiocyanate. Biochim Biophys Acta 1782:566–573
Tsuchihara K, Wazawa T, Ishii Y et al (2009) Characterization of chemoreceptive protein binding to an oviposition stimulant using a fluorescent micro-binding assay in a butterfly. FEBS Lett 583:345–349
Arlt VM, Wiessler M, Schmeiser HH (2000) Using polymerase arrest to detect DNA binding specificity of aristolochic acid in the mouse H-ras gene. Carcinogenesis 21:235–242
Kluck RM, Ellerby LM, Ellerby HM et al (2000) Determinants of cytochrome c pro-apoptotic activity. The role of lysine 72 trimethylation. J Biol Chem 275:16127–16133
Lartigue L, Kushnareva Y, Seong Y, Lin H, Faustin B, Newmeyer DD (2009) Caspase-independent mitochondrial cell death results from loss of respiration, not cytotoxic protein release. Mol Biol Cell 20:4871–4884
Yang NC, Ho WM, Chen YH, Hu ML (2002) A convenient one-step extraction of cellular ATP using boiling water for the luciferin-luciferase assay of ATP. Anal Biochem 306:323–327
Yuneva M, Zamboni N, Oefner P, Sachidanandam R, Lazebnik Y (2007) Deficiency in glutamine but not glucose induces MYC-dependent apoptosis in human cells. J Cell Biol 178:93–105
Acknowledgments
We wish to thank Dr. Yury Lazebnik (Cold Spring Harbor Laboratory) for his valuable recommendations and insights concerning our data. We would also like to thank members of the Chemical Biology group (Department of Pharmacology, Stony Brook University) for sharing their perspective on the data. This study was supported by the NIH grant P01 ES004068 to A. G.
Conflict of interest
The authors declare no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Romanov, V., Whyard, T., Bonala, R. et al. Glutamate dehydrogenase requirement for apoptosis induced by aristolochic acid in renal tubular epithelial cells. Apoptosis 16, 1217–1228 (2011). https://doi.org/10.1007/s10495-011-0646-5
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10495-011-0646-5