
Abstract
The death receptor ligand TRAIL is considered a promising candidate for cancer therapy because of its preferential toxicity to malignant cells. However its efficacy has been challenged by a number of resistance mechanisms. Therefore, agents that can overcome the resistance to enhance therapeutic efficacy of TRAIL are needed. In the current study, we found that bufalin, bufotalin and gamabufotalin, key members of bufadienolides isolated from a traditional Chinese medicine ChanSu, significantly potentiated human breast cancer cells with different status of ER-alpha to apoptosis induction of TRAIL, as evidenced by enhanced Annexin V/FITC positive cells (apoptotic cells), cytoplasmic histone-associated-DNA-fragments, membrane permeability transition (MPT), caspases activation and PARP cleavage. Further mechanistic investigation demonstrated that bufalin was able to significantly decrease Mcl-1 expression and modestly decrease Bcl-XL expression level. Down-regulations of these anti-apoptotic proteins were well correlated with inhibition of transcription factor STAT3 activation. The important consequence of down-regulation Mcl-1 in the enhancement action by combining bufalin with TRAIL was confirmed by either knockdown or overexpression of Mcl-1 approach. Our findings for the first time provided strong evidences that bufadienolide compounds have excellent potential to be developed as a novel class of sensitizers of TRAIL.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
One of the major drawbacks for conventional cancer therapeutic drugs is the indiscriminate killing of normal proliferative cells as well as cancer cells. Therefore agents that can selectively kill cancer cells without damage to healthy cells are much sought after to improve the specificity of cancer therapy. TNF-related apoptosis-inducing ligand (TRAIL) is a member of the TNF superfamily that induces apoptosis upon binding to its receptors (DR4, DR5) on the cell surface through the extrinsic caspase activation cascade involving DISC/procaspase-8 [1]. TRAIL preferentially kills cancer cells while leaving normal cells unscathed, thus it is considered as a promising novel anti-cancer biologic agent [1]. However, many types of cancer cells are resistant to TRAIL-induced apoptosis [2], posing major hurdle for its ultimately clinical utility. Several mechanisms have been proposed to explain TRAIL sensitivity/resistance among the various types of cancer cells [3, 4]. The first is defects in death receptor signaling (i.e., extrinsic pathway involving TRAIL/DR4/5/caspase-8), such as defects in the TRAIL receptors themselves or increased expression of inhibitors that are selective for death receptors such as FLIP or the decoy receptors DcR1 and DcR2. The second is presence of intracellular apoptosis inhibitory molecules that block death signaling downstream of death receptor activation and cross-talk with the intrinsic caspase activation pathway (i.e., mitochondria/cytochrome C/caspase-9/apoptosome), such as increased expression of caspase inhibitors cIAPs, survivin, or overexpression of Bcl-2 family anti-apoptotic proteins (Bcl-2, Bcl-XL and Mcl-1). The high frequency of TRAIL-resistant cancer cells requires strategies to sensitize them to TRAIL-induced apoptosis. One of the practical approaches to achieve this goal is to combine TRAIL with the agents that can reverse the resistance to restore sensitivity of cancer cells to TRAIL-induced apoptosis without an increase of toxicity towards normal cells [3, 4].
ChanSu, a traditional Chinese medicine, has been used to treat various diseases such as cardiac illness, pain, and cancer in China and other Asian countries [5, 6]. Bufadienolides are major class of biologically active compounds isolated from ChanSu [7]. The bufadienolides are a group of cardiotonic steroids [8, 9]. They first identified as a Na(+)/K(+)-ATPase inhibitory factor and considered to be important in the regulation of renal sodium transport and arterial pressure. More recent studies suggest that these compounds have the ability to regulate cell growth, differentiation, apoptosis and glucose metabolism [9]. Our previous study has shown that bufalin, bufotalin and gamabufotalin, three key members of bufadienolides were highly effective against BGC-823 human gastric cancer cells, Bel-7402 human hepatoma and HeLa human cervical cancer cells [10]. A growing body of literature documents that bufadienolides including bufalin and bufotalin can induce cell cycle arrest or apoptosis in multiple types of cancer cells, such as leukemia [11, 12], prostate [13], breast [14], lung [15, 16], liver [17], gastric [18] and ovarian cancer [19]. Apoptosis induction by bufadienolides was associated with mitochondrial pathway activation [13]. Furthermore, the in vivo efficacy of bufalin, the most well-studied bufadienolide, has been established in human cancer cell xenograft model in athymic nude mice [20] and a promising result has been obtained in a pilot human clinical study [6]. However, little is known about their potential use as an enhancer of anti-cancer biological or chemotherapeutic drugs. The objective of our current study was to examine the hypothesis that bufadienolides might sensitize cancer cells to TRAIL-induced apoptosis through facilitation of cross-talk with the mitochondrial caspase activation cascade.
We tested this hypothesis in human breast cancer cell models because many breast cancer cells are generally resistant to TRAIL-induced apoptosis [21]. We demonstrate here that bufalin, bufotalin and gamabufotalin, key members of bufadienolide compounds, greatly sensitize both estrogen receptor-alpha (ER-alpha)-positive MCF-7 and ER-alpha-negative MDA-MB-231 human breast cancer cells to TRAIL-induced apoptosis. Combining TRAIL with bufalin enabled TRAIL-induced death signaling to go through mitochondria to be amplified, which in turn led to enhanced caspase-dependent apoptosis. Because MDA-MB-231 and MCF-7 cells have constitutively activated STAT3, which increases transcription of several survival proteins including the Bcl-2 related molecule Mcl-1, we did further mechanistic studies that revealed a suppression of STAT3/Mcl-1 pathway by bufalin as one mechanism to enhance the apoptotic effects of TRAIL. To our knowledge this is the first report to suggest that bufadienolide compounds can be used as a potential enhancer of TRAIL efficacy against human breast cancer cells.
Materials and methods
Chemicals and reagents
TRAIL (SuperKillerTRAIL) was purchased from Alexis Biochemicals (San Diego, CA). Bufalin, bufotalin and gamabufotalin were isolated from Chinese Medicine ChanSu crude drug [10]. Antibodies specific for Bcl-2, Bcl-XL, Mcl-1, cleaved caspase-3, cleaved poly (ADP-ribose) polymerase (PARP; p89) and phospho-STAT3 were purchased from Cell Signaling Technology (Beverly, MA). Antibodies specific for caspase-8, -9, -7 and HA-tag were purchased from MBL International Corporation (Woburn, MA). An antibody for β-actin and JC-1 mitochondrial probe were purchased from Sigma (St. Louis, MO).
Cell culture and treatments
All cell lines used in this study, including MCF-7, MDA-MB-231 and MCF-10A, originated from the American Type Culture Collection. MCF-7 and MDA-MB-231 cells were grown in Dulbecco’s Modification of Eagle’s Medium (DMEM) supplemented with 10% fetal bovine serum without antibiotics. MCF-10A was grown in DMEM/F12K medium supplemented with 10 μg/ml insulin, 100 ng/ml cholera toxin, 5 mg/ml hydrocortisol and 20 ng/ml recombinant human epidermal growth factor. At 24–48 h after plating when cells were 50–60% confluence, the medium was changed before starting the treatment with bufadienolides and/or TRAIL.
Apoptosis evaluation
Apoptosis was assessed by multiple methods. The first was Annexin V staining of externalized phosphatidylserine in apoptotic cells by flow cytometry using Annexin V/FITC Staining Kit from MBL International. The second was a cell death detection ELISA kit to detect cytoplasmic histone-associated-DNA-fragments purchased from Roche Diagnostics. The third method was immunoblot analysis of PARP cleavage and caspase activation. All these methods were as described previously [22].
Determination of mitochondrial permeability transition (MPT)
Cells were treated with bufalin and/or TRAIL for 12 h, and then incubated with 1 ml medium containing 2 μg/ml JC-1 (5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolocarbocyanine iodide) for 20 min at 37°C in a humidified atmosphere containing 5% CO2. The stained cells were viewed and photographed under fluorescence microscope. For flow cytometry analysis, the treated cells were collected, and resuspended in 1 ml medium containing 2 μg/ml JC-1. After incubation at 37°C for 20 min, cells were analyzed by flow cytometry. The number of cells changing from red to green fluorescence indicates the frequency of cells exhibiting mitochondrial membrane depolarization.
Crystal violet staining
For the evaluation of overall inhibitory effect of bufalin and/or TRAIL on cell number, the cells were treated with bufalin and/or TRAIL for 24 h. After treatment, the culture medium was removed and the cells were fixed in 1% glutaraldehyde solution in phosphate buffered saline (PBS) for 15 min. The fixed cells were stained with 0.02% aqueous solution of crystal violet for 30 min. After washing with PBS, the stained cells were solubilized with 70% ethanol. The absorbance at 570 nm with the reference filter 405 nm was evaluated using a microplate reader (Thermo, USA).
Western blotting
The cell lysate was prepared in ice-cold radioimmuno-precipitation assay buffer. Cell lysates were resolved by electrophoresis and transferred to a PVDF membrane. The blot was then probed with primary antibody followed by incubation with the appropriate horseradish peroxidase-conjugated secondary antibodies. The signal was visualized by enhanced chemiluminescence (Pierce) and recorded on an X-ray film (Kodak).
RNA interference
The siRNA for human Mcl-1 and non-targeting siRNA were purchased from Santa Cruz Biotechnologies (Santa Cruz, CA). The cells were transfected with 50 nmol/l siRNAs using siPORT™ NeoFX™ Transfection Agent (Ambion, Austin, TX) for 24 h and then were used for subsequent experiments with TRAIL treatment.
Transient transfection
MDA-MB-231 cells were transiently transfected with pCMV-HA-Mcl-1 plasmid [23] (generously provided by Carine Michiels, University of Namur) or control plasmid with the use of Lipofectamine 2000 (Invitrogen). To monitor the transfection efficiency, cells were transfected in parallel with green fluorescent protein plasmids (pGFP-C1; Clontech). The transfection efficiency was ~60–70%.
Statistical analysis
Data are presented as mean ± SE. A one-way ANOVA was used for comparison of multiple groups. A t-test was used for comparison of two groups. Statistical difference was set at P values of <0.05.
Results
Bufadienolide compounds greatly sensitized ER-alpha-positive MCF-7 and ER-alpha-negative MDA-MB-231 human breast cancer cells to TRAIL-induced apoptosis
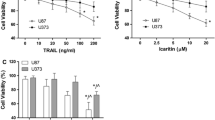
In order to increase the likelihood of detecting an enhanced combination response, we chose doses of bufadienolide compounds or TRAIL that by themselves would only minimally or modestly induced apoptosis in MCF-7 and MDA-MB-231 cells based on our preliminary dose-range finding experiments. As shown in Fig. 1a, the percentage of apoptotic cells (Annexin V/FITC positive cells) at 24 h in MCF-7 cells was 3.5 ± 0.18% in control cells, 4.0 ± 0.87% with 0.02 μmol/l bufalin and 11.7 ± 0.47% with 50 ng/ml TRAIL. In contrast, combining bufalin with TRAIL led to a dramatic increase of apoptotic cells percentage (48.7 ± 9.3%, P < 0.01 compared with sum of single agent treatment). To determine whether the enhancement action is specific for ER-alpha-positive MCF-7 cells, we measured apoptosis induction by the combination in ER-alpha-negative MDA-MB-231 cells. As shown in Fig. 1b, bufalin treatment alone for 24 h caused a slight increase of apoptosis level (4.2 ± 0.35% in bufalin-treated cells vs 3.7 ± 0.03% in control cells) while TRAIL alone induced a modest increase of apoptosis (8.5 ± 1.2% in TRAIL-treated cells vs 3.7 ± 0.03% in control cells) in MDA-MB-231 cells. Similar to MCF-7 cells, the bufalin/TRAIL combination treatment caused a significantly enhanced apoptotic effect in MDA-MB-231 cells (31.5 ± 5.8%, P < 0.01 compared with sum of single agent treatment). These results suggest that bufalin can sensitize both ER-alpha (−) and ER-alpha (+) breast cancer cells to TRAIL-induced apoptosis.

Bufadienolide compounds sensitize human breast cancer cells to TRAIL-induced apoptosis. MCF-7, MDA-MB-231 and MCF-10A cells were treated with bufadienolide compounds, TRAIL or bufadienolide compounds/TRAIL combination at the indicated concentrations for 24 h. The apoptosis induction was evaluated by Annexin V/FITC apoptosis detection kit or a cell death ELISA kit. a apoptosis induction by TRAIL and/or bufalin in MCF-7 cells, b apoptosis induction by TRAIL and/or bufalin in MDA-MB-231 cells, c apoptosis induction by TRAIL and/or bufotalin or gamabufotalin in MCF-7 cells, d apoptosis induction by TRAIL and/or bufotalin or gamabufotalin in MDA-MB-231 cells, e apoptosis induction by TRAIL and/or bufalin in MCF-10A cells. n = 3, P < 0.01
To determine whether the enhancement action can be also achieved by other bufadienolide compounds, we assessed apoptosis induction by combining TRAIL with bufotalin and gamabufotalin, the other two key members of bufadienolides in MCF-7 and MDA-MB-231cells by a cell death ELISA kit (Roche) or annexin V staining as described in “Materials and methods” section respectively. As shown in Fig. 1c, each single treatment induced a slight increase of apoptosis while the combinations led to a significant increase of apoptosis by ~threefold over the sum of single agents in MCF-7 cells. Similar enhancement action was also found in MDA-MB-231 cells (Fig. 1d). These results suggest general applications of the sensitization action by bufadienolide compounds.
To determine whether the enhancement action is specific toward cancer cells, we examined apoptosis induction by the combination in MCF-10A, a non-tumorigenic immortalized human breast epithelial cell line. As shown in Fig. 1e, bufalin (0.25 μM) or TRAIL (100 ng/ml) alone treatment caused a minimal apoptosis (4.5 ± 1.9% in bufalin-treated cells, 7.5 ± 0.7% in TRAIL-treated cells vs 3.4 ± 0.4% in control cells) while the combination led to only a slight further increase of apoptosis (11.3 ± 1.5%) over the agent alone treatment in MCF-10A cells. Similar patterns of apoptosis induction were found in bufotalin or gamabufotalin combining with TRAIL in MCF-10A cells (data not shown). Taken together, bufadienolide compounds can preferentially enhance TRAIL-induced apoptosis in human breast cancer cells without apparent increase of toxicity in normal breast epithelial cells.
Enhanced apoptotic effects by TRAIL/bufalin combo were associated with augmentation of caspases activation
As mentioned above, TRAIL-resistance can occur at the level of death receptor activation and/or its downstream caspase activation cascade and cross-talk to intrinsic pathway [3, 4]. Given MCF-7 and MDA-MB-231 cells express death receptor DR4 and DR5 [24], we postulated that the sensitization might take place at the level of mitochondrial activation. To test this hypothesis, we examined caspases activation by western blotting analysis of caspases expression or cleavages. As shown in Fig. 2a, TRAIL alone induced a significant cleavage of caspase-8, but without apparent cleavage of caspase-9, suggesting TRAIL-induced death signaling failed to cross-talk with mitochondrial pathway for efficient amplification in MDA-MB-231 cells. TRAIL alone induced a modest cleavage of caspase-3, which was likely due to caspase-8 direct effect. Neither caspase-8 nor caspase-9 was activated by bufalin alone treatment. Combining TRAIL with bufalin induced a further cleavage of caspase-8. More importantly, the combination caused a significant cleavage of caspase-9 (mitochondrial activation), which in turn led to a strongly enhanced activation of caspase-3 and apoptosis. Similar patterns of caspases activation were found in MCF-7 cells (Fig. 2b). To further confirm the enhanced mitochondrial activation by the combination, the mitochondrial membrane potential, a direct indicator of mitochondrial activation, was measured by fluorescence microscope and flow cytometry following JC-1 mitochondrial probe staining as described in “Materials and methods” section. As shown in Fig. 2c, the percentage of green fluorescence-positive cells (representing mitochondrial membrane–depolarized cells) at 12 h was 7.9 ± 0.54% in control cells, 8.9 ± 0.55% with bufalin, 12.6 ± 1.81% with TRAIL and 47 ± 4.83% in cells treated with combining bufalin with TRAIL in MCF-7 cells (P < 0.01 compared with sum of TRAIL and bufalin alone treatment). Similar patterns of mitochondrial activation by bufalin and/or TRAIL were found in MDA-MB-231 cells (Fig. 2d). These mitochondrial membrane potential changes were well correlated with apoptosis induction by the combination. To determine the role of caspases activation in apoptosis induction by the combination, we tested the effect of a pan-caspase inhibitor on apoptosis induced by the combination treatment. As shown in Fig. 2e, inhibition of caspases activation completely blocked apoptosis induction by the combined treatment. Taken together, these results suggest that combining TRAIL with bufalin enabled TRAIL-induced death signaling to go through mitochondria resulting in fully activation of the extrinsic and intrinsic caspase pathways and activation of caspases was critical for apoptosis induction by the combination.

Enhanced activation of caspases by bufalin/TRAIL combination. The cells were treated with bufalin, TRAIL or bufalin/TRAIL combination for 24 h and western blotting was used to analyze expression or cleavage of caspase-8, -9 and caspase-3/-7 in MDA-MB-231 (a) and MCF-7 cells (b). Disruption of mitochondrial membrane potential by bufalin/TRAIL combination. The cells were treated with bufalin, TRAIL or bufalin/TRAIL combination for 12 h and stained with JC-1 mitochondrial probe. The stained cells were viewed and photographed under inverted microscope and quantitatively analyzed by flow cytometry c MCF-7 cells, d MDA-MB-231 cells. e Effects of pan-caspase inhibitor on apoptosis induction by TRAIL/bufalin combination in MDA-MB-231 cells. n = 3, P < 0.01
Down-regulation of Mcl-1 expression by bufalin
Mitochondrial intrinsic caspase activation cascade is tightly controlled by Bcl-2 family proteins [25]. Especially anti-apoptotic Bcl-2 family proteins have been shown to involve in TRAIL-resistance [3, 4]. To further determine the molecular targets that contributed to promotion of mitochondrial activation by bufalin, we analyzed the expression levels of anti-apoptotic Bcl-2 family proteins Bcl-2, Bcl-XL and Mcl-1. As shown in Fig. 3a, bufalin treatment caused a dramatic decrease of Mcl-1 protein level in a concentration-dependent manner in both MCF-7 and MDA-MB-231 cells. Exposure to 0.25 μmol/l bufalin led to significant decrease of Mcl-1 abundance as early as 12 h (Fig. 3b), and persisted through 72 h (data not shown) in MDA-MB-231 cells. Bufalin treatment also induced a modest decrease of Bcl-XL abundance without affecting Bcl-2 protein level (Fig. 3a). The results suggest that Mcl-1 might be a potential key target for bufalin to facilitate mitochondrial activation.

Downregulation of anti-apoptotic protein Mcl-1 expression by bufalin. a Bufalin inhibited Mcl-1 expression in a concentration-dependent manner in MCF-7 and MDA-MB-231 cells, b time course of Mcl-1 expression inhibition by bufalin in MDA-MB-231 cells, c inhibition of Mcl-1 expression by bufalin/TRAIL combination in MDA-MB-231 cells
Inhibition of constitutive activation of STAT3 by bufalin
Both MDA-MB-231 and MCF-7 cells have constitutively active STAT3 [26, 27], which activates the transcription of Mcl-1 and Bcl-XL [28], we therefore hypothesize that suppression of STAT3 activation might contribute to Mcl-1 and Bcl-XL transcriptional inhibition which in turn leads to their protein level reduction. Phosphorylation of STAT3 at Tyr705 was examined by western blotting analysis using specific phospho-STAT3 antibody. As we expected, bufalin treatment caused concentration and time-dependent inhibition of STAT3 activation (phosphorylation) in the MDA-MB-231 cells. Exposure to 0.25 μmol/l bufalin led to a rapid (as early as 6 h) and persistent (throughout 24 h) inhibition of STAT3 Tyr705 phosphorylation (Fig. 4b), which was 6 h ahead of its transcriptional target Mcl-1 protein change. These results indicate that inhibition of constitutively active STAT3 was likely one mechanism underlying down-regulation of Mcl-1 and Bcl-XL by bufalin.

Inhibition of constitutive activation of STAT3 by bufalin in MDA-MB-231 cells. a Bufalin inhibited STAT3 Tyr705 phosphorylation in a concentration-dependent manner, b time course of STAT3 phosphorylation inhibition by bufalin
Knockdown of Mcl-1 sensitized MDA-MB-231 cells to TRAIL-induced apoptosis and overexpression of Mcl-1 attenuated apoptosis induction by TRAIL/bufalin combo treatment
To critically test the functional significance of downregulation of Mcl-1 in the sensitization of breast cancer cells to TRAIL, we first knocked down Mcl-1 to examine the effects of down-regulation of Mcl-1 on TRAIL-induced apoptosis in MDA-MB-231 cells. As shown in Fig. 5a by western blotting detection, Mcl-1 siRNA caused a significant reduction of Mcl-1 protein expression, but did not increase PARP cleavage and apoptotic DNA fragmentation in comparison with non-targeting siRNA control, measured by western blotting and a Cell Death ELISA kit respectively (Fig. 5a, b). However, downregulation of Mcl-1 by RNAi significantly increased TRAIL-induced PARP cleavage and apoptotic DNA fragmentation (0.52 ± 0.02 in non-targeting siRNA control cells vs 1.1 ± 0.02 in Mcl-1 siRNA cells, P < 0.01), suggesting downregulation of Mcl-1 is sufficient to sensitize MDA-MB-231 cells to TRAIL-induced apoptosis.

Inhibition of Mcl-1 expression by RNAi is sufficient to sensitize MDA-MB-231 cells to TRAIL-induced apoptosis. a Knockdown efficiency and PARP cleavage were examined by western blotting, b apoptosis induction was assessed by a cell death ELISA kit to determine effects of down-regulation of Mcl-1 expression on TRAIL sensitivity. n = 3, P < 0.01
We next used overexpression of Mcl-1 method to determine the effects of up-regulation of Mcl-1 expression on combining bufalin with TRAIL-induced apoptosis in MDA-MB-231 cells. MDA-MB-231 cells were transiently transfected with an expression vector containing the fulllength Mcl-1 cDNA in-frame with an HA-tag (pHA-Mcl-1) as described in “Materials and methods” section. The overexpressed exogenous Mcl-1 was checked using western blotting analysis of HA-tag or HA-Mcl-1 (Fig. 6a). Overexpression of Mcl-1 led to ~40% reduction of apoptosis by the combination in comparison with vector control (Fig. 6b, 1.63 ± 0.1 vs. 1.01 ± 0.09, P < 0.05). Together, these results suggest that Mcl-1 is a likely molecular target for sensitizing these breast cancer cells to TRAIL-induced apoptosis by TRAIL combining with bufalin.

Overexpression of Mcl-1 significantly attenuates bufalin/TRAIL combination-induced apoptosis in MDA-MB-231 cells. a Exogenous Mcl-1 (HA-Mcl-1) expression level was examined by western blotting, b apoptosis induction was assessed by a cell death ELISA kit to determine effects of up-regulation of Mcl-1 expression on bufalin/TRAIL combination-induced apoptosis. n = 3, P < 0.05
Discussion
TRAIL-based combinatorial therapy is considered an attractive approach to tackle its resistance problem [3]. In the present study, we found that combining TRAIL with bufalin, bufotalin or gamabufotalin, key members of bufadienolide compounds, led to a strong enhancement of apoptotic effects in two human breast cancer cells with different status of ER-alpha. MDA-MB-231 is a typical example of triple negative breast cancer that does not have effective therapeutic options [29]. Our data strongly suggest potential usefulness of bufadienolide compounds as a novel class of TRAIL-sensitizing agents, especially for the triple negative breast cancer.
It has been shown that many human breast cancer cells are highly resistant to TRAIL-induced apoptosis [21]. Our apoptosis evaluation results show that 100 or 50 ng/ml TRAIL induced a minimal apoptosis after 24 h treatment in MDA-MB-231 cells (Fig. 1b) and a modest apoptosis in MCF-7 cells (Fig. 1a), respectively, even though MDA-MB-231 cells are considered to be sensitive to TRAIL in some reports [30]. Nevertheless, combining sub-lethal dose of bufalin with TRAIL led to a greater apoptosis induction in both ER-alpha-negative MDA-MB-231 and ER-alpha-positive MCF-7 human breast cancer cells. These enhanced apoptotic effects further led to a greater reduction of cell number than the sum achieved by each single agent (data not shown), suggesting improvement of therapeutic efficacy is likely to be achieved by bufalin/TRAIL combination.
Furthermore, this apoptosis enhancement action was applicable to bufotalin and gamabufotalin, the other two bufadienolides. More importantly, this enhancement effect was specific for breast cancer cells without increasing toxicity to normal breast epithelial cells, adding a desirable feature of bufadienolide compounds as a novel class of TRAIL sensitizers.
Most human breast cancer cells including MDA-MB-231 and MCF-7 cells express death receptors DR4 and DR5 [24], therefore the TRAIL-resistance in breast cancer cells is likely mediated by defects in the signaling pathways downstream of death receptor activation and cross-talk with mitochondrial pathway activation. Indeed in TRAIL-resistance cells, TRAIL-induced extrinsic death signaling was often stuck at mitochondrial level [31–33]. The results of caspases activation analysis demonstrated that TRAIL alone treatment can trigger activation of caspase-8 but not caspase-9 cascade in both MDA-MB-231 and MCF-7 cells (Fig. 2a, b). Combining TRAIL with bufalin resulted in activation of not only death receptor pathway, but also mitochondrial pathway, suggesting that bufalin treatment enabled TRAIL-induced death signaling to cross-talk with mitochondrial pathway for efficient amplification. Our data therefore support our hypothesis that bufalin sensitized breast cancer cells to TRAIL through promotion of mitochondrial pathway activation.
As alluded to in the Introduction, overexpression of mitochondrial membrane potential controller Bcl-2 family antiapoptotic proteins in cancer cells has been recognized as an important mechanism of developing resistance to TRAIL [3, 4]. Mcl-1, a member of anti-apoptotic Bcl-2 family proteins, has been found overexpressed in many types of cancer including breast [34]. Recent studies suggest that Mcl-1 is one of the key targets to sensitize cancer cells to TRAIL-induced apoptosis [35]. In the present study, we demonstrated that bufalin decreased Mcl-1 protein level dramatically and persistently, suggesting its involvement in the enhancement action by bufalin. To confirm biological consequence of downregulation Mcl-1 expression, we manipulated Mcl-1 protein level by either RNAi or overexpression method to test effects of Mcl-1 protein levels on TRAIL or combination-induced apoptosis in MDA-MB-231 cells. It has been shown that inhibition of Mcl-1 expression is both necessary and sufficient for apoptosis induction in some systems whereas this event is not the only hit needed for apoptosis in other systems [36]. Our Mcl-1 knockdown experiment with siRNA showed that inhibition of Mcl-1 is not sufficient to induce apoptosis in MDA-MB-231 cells (Fig. 5a, b), which is consistent with the observation that bufalin caused a significant inhibition of Mcl-1 expression without apoptosis in MDA-MB-231 cells. However, knockdown of Mcl-1 is sufficient to sensitize MDA-MB-231 cells to TRAIL-induced apoptosis (Fig. 5a), providing evidence for Mcl-1 as a reasonable target for sensitization of breast cancer cells to TRAIL. Our Mcl-1 overexpression experiment further confirmed that downregulation of Mcl-1 is required for bufalin to sensitize MDA-MB-231 cells to TRAIL (Fig. 6b). Regarding the mechanism of Mcl-1 downregulation by bufalin, we identified that suppression of constitutive activation of STAT3, a well known transcriptional factor of Mcl-1, might contribute to Mcl-1 transcriptional inhibition, providing a possible mechanistic explanation for down-regulation of Mcl-1 by bufalin (Fig. 4).
In summary, bufadienolide compounds can greatly sensitize both ER-alpha-negative MDA-MB-231 and ER-alpha-positive MCF-7 human breast cancer cells to TRAIL-induced apoptosis without increased toxicity to normal breast epithelial cells. We also revealed that the sensitization effects of bufalin are mediated at least by down-regulation of STAT3/Mcl-1, key negative regulators of TRAIL-induced apoptosis. Our findings provided the first evidence that bufadienolide compounds might be a novel class of TRAIL-sensitizing agents through enhancing cross-talk between apoptosis pathways (Fig. 7).

Signaling pathway underlying TRAIL and/or bufalin-induced apoptosis in breast cancer cells. TRAIL induces caspase-8 activation, whereas bufalin induces down-regulation of the pro-survival mitochondrial proteins. Combining TRAIL with bufalin enabled TRAIL-induced death signaling to go through mitochondria to be amplified, which in turn led to enhanced apoptosis
References
Ashkenazi A, Dixit VM (1998) Death receptors: signaling and modulation. Science 281:1305–1308
Shankar S, Srivastana RK (2004) Enhancement of therapeutic potential of TRAIL by cancer chemotherapy and irradiation: mechanisms and clinical implications. Drug Resist Updat 7(2):139–156
Thorburn A, Behbakht K, Ford H (2008) TRAIL Receptor-targeted therapeutics: resistance mechanisms and strategies to avoid them. Drug Resist Updat 11(1–2):17–24
Kim SH, Ricci MS, El-Deiry WS (2008) Mcl-1: a gateway to TRAIL sensitization. Cancer Res 68(7):2062–2064 Review
Gao H, Zehl M, Kaehlig H, Schneider P, Stuppner H, Moreno Y et al (2010) Rapid structural identification of cytotoxic bufadienolide sulfates in toad venom from Bufo melanosticus by LC-DAD-MS(n) and LC-SPE-NMR. J Nat Prod 73(4):603–608
Meng Z, Yang P, Shen Y, Bei W, Zhang Y, Ge Y et al (2009) Pilot study of huachansu in patients with hepatocellular carcinoma, nonsmall-cell lung cancer, or pancreatic cancer. Cancer 115(22):5309–5318
Nogawa T, Kamano Y, Yamashita A, Pettit GR (2001) Isolation and structure of five new cancer cell growth inhibitory bufadienolides from the Chinese traditional drug Ch’an Su. J Nat Prod 64(9):1148–1152
Puschett JB, Agunanne E, Uddin MN (2010) Emerging role of the bufadienolides in cardiovascular and kidney diseases. Am J Kidney Dis 56(2):359–370
Bagrov AY, Shapiro JI, Fedorova OV (2009) Endogenous cardiotonic steroids: physiology, pharmacology, and novel therapeutic targets. Pharmacol Rev 61(1):9–38
Ye M, Guo H, Guo H, Han J, Guo D (2006) Simultaneous determination of cytotoxic bufadienolides in the Chinese medicine ChanSu by high-performance liquid chromatography coupled with photodiode array and mass spectrometry detections. J Chromatogr B 838(2):86–95
Jing Y, Ohizumi H, Kawazoe N, Hashimoto S, Masuda Y, Nakajo S et al (1994) Selective inhibitory effect of bufalin on growth of human tumor cells in vitro: association with the induction of apoptosis in leukemia HL-60 cells. Jpn J Cancer Res 85(6):645–651
Watabe M, Masuda Y, Nakajo S, Yoshida T, Kuroiwa Y, Nakaya K (1996) The cooperative interaction of two different signaling pathways in response to bufalin induces apoptosis in human leukemia U937 cells. J Biol Chem 271(24):14067–14072
Yu CH, Kan SF, Pu HF, Jea Chien E, Wang PS (2008) Apoptotic signaling in bufalin- and cinobufagin-treated androgen-dependent and -independent human prostate cancer cells. Cancer Sci 99(12):2467–2476
Winnicka K, Bielawski K, Bielawska A, Surazyński A (2008) Antiproliferative activity of derivatives of ouabain, digoxin and proscillaridin A in human MCF-7 and MDA-MB-231 breast cancer cells. Biol Pharm Bull 31(6):1131–1140
Pan W, Qu J, Chen T, Sun L, Qi J (2009) FLIM and emission spectral analysis of caspase-3 activation inside single living cell during anticancer drug-induced cell death. Eur Biophys J 38(4):447–456
Wang J, Jin Y, Xu Z, Zheng Z, Wan S (2009) Involvement of caspase-3 activity and survivin downregulation in cinobufocini-induced apoptosis in A 549 cells. Exp Biol Med (Maywood) 234(5):566–572
Su CL, Lin TY, Lin CN, Won SJ (2009) Involvement of caspases and apoptosis-inducing factor in bufotalin-induced apoptosis of Hep 3B cells. J Agric Food Chem 57(1):55–61
Li D, Qu X, Hou K, Zhang Y, Dong Q, Teng Y et al (2009) PI3K/Akt is involved in bufalin-induced apoptosis in gastric cancer cells. Anticancer Drugs 20(1):59–64
Takai N, Ueda T, Nishida M, Nasu K, Narahara H (2008) Bufalin induces growth inhibition, cell cycle arrest and apoptosis in human endometrial and ovarian cancer cells. Int J Mol Med 21(5):637–643
Han KQ, Huang G, Gu W, Su YH, Huang XQ, Ling CQ (2007) Anti-tumor activities and apoptosis-regulated mechanisms of bufalin on the orthotopic transplantation tumor model of human hepatocellular carcinoma in nude mice. World J Gastroenterol 13(24):3374–3379
Rahman M, Pumphrey JG, Lipkowitz S (2009) The TRAIL to targeted therapy of breast cancer. Adv Cancer Res 103:43–73 (Review)
Hu H, Jiang C, Schuster T, Li GX, Daniel PT, Lü J (2006) Inorganic selenium sensitizes prostate cancer cells to TRAIL-induced apoptosis through superoxide/p53/Bax-mediated activation of mitochondrial pathway. Mol Cancer Ther 5(7):1873–1882
Piret JP, Minet E, Cosse JP, Ninane N, Debacq C, Raes M et al (2005) Hypoxia-inducible factor-1-dependent overexpression of myeloid cell factor-1 protects hypoxic cells against tert-butyl Hydroperoxide-induced apoptosis. J Biol Chem 280(10):9336–9344
Keane MM, Ettenberg SA, Nau MM, Russell EK, Lipkowitz S (1999) Chemotherapy augments TRAIL-induced apoptosis in breast cell lines. Cancer Res 59:734–741
Green DR (2006) At the gates of death. Cancer Cell 9(5):328–330
Garcia R, Bowman TL, Niu G, Yu H, Minton S, Muro-Cacho CA et al (2001) Constitutive activation of Stat3 by the Src and JAK tyrosine kinases participates in growth regulation of human breast carcinoma cells. Oncogene 20:2499–2513
Evans MK, Yu CR, Lohani A, Mahdi RM, Liu X, Trzeciak AR et al (2007) Expression of SOCS1 and SOCS3 genes is differentially regulated in breast cancer cells in response to proinflammatory cytokine and growth factor signals. Oncogene 26(13):1941–1948
Buettner R, Mora LB, Jove R (2002) Activated STAT signaling in human tumors provides novel molecular targets for therapeutic intervention. Clin Cancer Res 8(4):945–954 (Review)
Ray M, Polite BN (2010) Triple-negative breast cancers: a view from 10,000 feet. Cancer J 16(1):17–22 (Review)
Rahman M, Davis SR, Pumphrey JG, Bao J, Nau MM, Meltzer PS et al (2009) TRAIL induces apoptosis in triple-negative breast cancer cells with a mesenchymal phenotype. Breast Cancer Res Treat 113(2):217–230
Liang Y, Eid MA, Lewis RW, Kumar MV (2005) Mitochondria from TRAIL-resistant prostate cancer cells are capable of responding to apoptotic stimuli. Cell Signal 17:243–251
Ravi R, Fuchs EJ, Jain A, Pham V, Yoshimura K, Prouser T et al (2006) Resistance of cancers to immunologic cytotoxicity and adoptive immunotherapy via X-linked inhibitor of apoptosis protein expression and coexisting defects in mitochondrial death signaling. Cancer Res 66(3):1730–1739
Ndozangue-Touriguine O, Sebbagh M, Mérino D, Micheau O, Bertoglio J, Bréard J (2008) A mitochondrial block and expression of XIAP lead to resistance to TRAIL-induced apoptosis during progression to metastasis of a colon carcinoma. Oncogene 27(46):6012–6022
Hsieh FC, Cheng G, Lin J (2005) Evaluation of potential Stat3-regulated genes in human breast cancer. Biochem Biophys Res Commun 335(2):292–299
Ricci MS, Kim SH, Ogi K, Plastaras JP, Ling J, Wang W et al (2007) Reduction of TRAIL-induced Mcl-1 and cIAP2 by c-Myc or sorafenib sensitizes resistant human cancer cells to TRAIL-induced death. Cancer Cell 12(1):66–80
Derenne S, Monia B, Dean NM, Taylor JK, Rapp MJ, Harousseau JL et al (2002) Antisense strategy shows that Mcl-1 rather than Bcl-2 or Bcl-x(L) is an essential survival protein of human myeloma cells. Blood 100(1):194–199
Acknowledgments
This work was supported by grants from Chinese Universities Scientific Fund (2009-2-11), National Natural Science Foundation of China (NSFC, 31071533, 30972172). We thank Prof. Carine Michiels for generously providing the pCMV-HA-Mcl-1 plasmid and Prof. Junxuan Lu for providing valuable comments and writing assistance.
Conflict of interest
The authors have no conflicts of interest to disclosure.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article
Cite this article
Dong, Y., Yin, S., Li, J. et al. Bufadienolide compounds sensitize human breast cancer cells to TRAIL-induced apoptosis via inhibition of STAT3/Mcl-1 pathway. Apoptosis 16, 394–403 (2011). https://doi.org/10.1007/s10495-011-0573-5
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10495-011-0573-5