
Abstract
Apoptosis and oncotic necrosis in neuronal and glial cells have been documented in many neurological diseases. Distinguishing between these two major types of cell death in different neurological diseases is needed in order to better reveal the injury mechanisms so as to open up opportunities for therapy development. Accumulating evidence suggests apoptosis and oncosis epitomize the extreme ends of a broad spectrum of morphological and biochemical events. Biochemical markers that can distinguish between the calpain and caspase dominated types of cell death would help in this process. In this study, three chemical agents, maitotoxin (MTX), staurosporine (STS) and thylenediaminetetraacetic acid (EDTA), were used to induce different types of cell death in PC12 neuronal-like cells. MTX-induced necrosis, as determined by the increased levels of calpain-specific cleaved fragments of spectrin by antibodies specific to the calpain-cleaved 150 kDa αII-spectrin breakdown product (SBDP150) and 145 kDa αII-spectrin breakdown product (SBDP145). In this paradigm, there were no detectable SBDP150i and SBDP120 fragments as determined by antibodies specific to the caspase-cleaved specific fragments similar to those seen in the EDTA-mediated apoptotic PC-12 cells. In contrast to the calpain specific MTX necrosis treatment and the caspase EDTA apoptotic treatment is the STS treatment which induced both proteases as shown by the increase in all the SBDP fragments. Furthermore, compared to SBDP150, SBDP145 appears to be a more specific and sensitive biomarker for calpain activation. Taken together, our results suggested calpains and caspases which dominate the two major types of cell death could be independently discriminated by specifically examining the multiple αII-spectrin cleavage breakdown products.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Programmed cell death (PCD) in neuronal and glial cells has been documented in many neurological diseases, such as traumatic brain injury (TBI), stroke and Alzheimer’s disease [1–4]. Generally oncotic necrosis of neurons has been identified in the acute post-traumatic period within contused or hemorrhaging regions while the apoptotic neurons were localized in regions further from the site of impact in the days and weeks after trauma [1, 5, 6]. The ability to distinguish the two major types of cell death (oncotic necrosis and apoptosis) in different neurological diseases should provide improved insight into the injury mechanisms of each specific pathoneurological condition. This can also help better diagnose patients and better pinpoint targets for therapy development [7–9].
Traditionally, morphological studies, including triphenyltetrazolium chloride (TTC), hematoxylin and eosin (H&E), and terminal deoxynucleotidyl transferase mediated dUTP nick end labeling (TUNEL) staining have been used to address the role of apoptosis versus necrosis. Oncotic necrosis, from the Greek meaning “swelling and degenerating corpse,” is characterized by cell and organelle swelling leading to nuclear degradation and, ultimately, to disruption of the cell membrane and cell lysis [10, 11]. It has been shown that when these rapid and active molecular and biochemical processes occur there is an increase in the activation of members of the proteolytic calpain family [7, 12].
Apoptosis, on the other hand, is characterized by a more well-defined series of morphological and biochemical traits. It typically includes the activation of a specific family of proteases known as caspases [13], although caspase-independent processes involving proteins such as AIF have been reported [14, 15]. Kerr et al. [16] first characterized this mode of cell death and labeled it “apoptosis”, from the Greek meaning “falling leaves”, to emphasize the cells’ demise into small membrane bound particles. The features of apoptosis include chromatin condensation, internucleosomal DNA fragmentation, cell shrinkage and finally cell dismantlement into membrane-enclosed vesicles [17].
Accumulating evidence suggests apoptosis and oncosis epitomize the opposite ends of a broad spectrum of morphological and biochemical events. However under certain stress conditions neither oncosis nor apoptosis may predominate suggesting that a mixture of the two may occur either simultaneously or along a time scale. The cross-talk between the two cysteine protease systems involved in PCD, calpains and caspases, further blur the boundary between apoptosis and necrosis [18–22]. Furthermore, as more has become known about these two cell death processes morphological change can no longer be considered the most practical end-point measurement thus restricting its usefulness for clinical studies [23].
Finding biochemical markers which can distinguish between calpain and caspase dominated types of cell death is needed and has the potential of monitoring the endpoints of the therapeutic progress.
Recently, the degradation of αII-spectrin as a useful biomarker for the level and mechanism of injury has been documented in TBI, stroke and aneurysmal subarachnoid hemorrhage. [2, 24–27]. AlphaII-spectrin, a 280 kDa cytoskeletal protein found on the intracellular side of the plasma membrane, forms part of the membrane scaffolding that is cross linked with actin [28] thereby providing a cell with structure [29, 30]. In certain types of brain injury, such as diffuse axonal injury due to TBI, spectrin will be irreversibly cleaved by a member of one of two families of proteolytic enzymes, either calpains or caspases [31, 32].
A number of drawbacks have, until recently, limited the usage of total αII-spectrin degradation as a biomarker to distinguish calpain from caspase dominant necrotic and apoptotic neuronal cell death. For example, it is difficult to distinguish the calpain and caspase-generated SBDP150 and SBDP145 on the western blot membranes unless they are completely separated by SDS–PAGE gel electrophoresis [33–38]. Also, intact αII-spectrin lacks calpain and caspase dominant cell death specificity when used in ELISA endpoint measurements. Finally, calpain and caspase-3 each activation generate one αII-spectrin fragment at 150 kDa so that SBDP150 is often mistakenly considered to be a non-specific spectrin breakdown product (SBDP) [30].
In this study, we examined αII-spectrin and the cleavage products for both calpains (necrosis and apoptosis) and caspases (apoptosis) following treatment by various chemical agents. We demonstrated here that the two cell death pathways could be independently demonstrated by examining the αII-spectrin cleavage breakdown products. In the process of our study, a calpain-mediated SBDP150 and caspase-3 SBDP150 (SBDP150i) products could be distinguished using fragmental-specific antibodies. Lastly, our results suggest SBDP145 is a more specific and sensitive biomarker for calpain activation than SBDP150.
Materials and methods
BDP-specific antibodies
Antibody specific to calpain-generated αII-spectrin fragment SBDP150N (raised against new C-terminal QQQEV-COOH), SBDP150 (raised against new N-terminal NH2-GMMPR), SBDP145 (raised against new N-terminal NH2-SAHEV) and caspase-generated SBDP120 (raised against NH2-SVEAL) as well as SBDP150i (NH2-SKTAS) were synthesized. Briefly, custom-made peptides (California Peptide, Napa, CA) were coupled via C-terminal cysteine [C] to Keyhole Limpet Hemocyanin (KLH) protein using a sulfo-link crosslinking reagent (Pierce). The conjugated peptide (2 mg) was injected into two rabbits. After 3 months, serum samples from the rabbits were collected. The affinity-purified antibody was dialyzed against TBS (20 mM Tris–HCl, pH 7.4, 150 mM NaCl), concentrated and stored in 50% glycerol at −20°C.
Rat PC12 pheochromocytoma cell culture
Rat PC12 cells were grown on polystyrene tissue culture dishes (Falcon, Becton–Dickinson, Franklin Lakes, NJ, USA) in Dulbecco’s modified Eagle’s medium (Gibco, Invitrogen Corp., Grand Island, NY, USA) supplemented with 5% horse serum (Gibco), 1% Fungizone (Gibco), 100 units/ml penicillin and 100 μg/ml streptomycin (Gibco) and kept at 37°C in a humidified 5% CO2 incubator for 12–24 h before treatment.
Cell collection and preparation for immunoblots
The PC12 cells were treated for various time periods, washed twice with phosphate buffered saline (PBS), and solubilized with lysis buffer containing 20 mM Tris–HCl, pH 7.4, 150 mM NaCl, 5 mM EDTA, 5 mM EGTA, 1% Triton X-100, 1 mM NaF, 1 mM Na3VO4, and a protease inhibitor cocktail tablet (Roche, Indianapolis, IN). Samples (20 μg of protein) were resolved by Tris/glycine gel (Invitrogen Life Technologies, Carlsbad, CA, USA) sodium dodecyl sulfate–polyacrylamide electrophoresis (SDS–PAGE) and transferred onto a polyvinylidene fluoride (PVDF) membrane (Bio-Rad Laboratories) by the semidry method. Membranes were incubated with the primary antibody (mouse monoclonal or rabbit polyclonal) in 5% non-fat milk in TBST at 4°C overnight. Following a series of washes with TBST, membranes were incubated for one hour at room temperature with a biotinylated secondary antibody (horseradish peroxidase-conjugated goat anti-mouse IgG or goat anti-rabbit IgG—1:5,000 dilution). Following another series of washes, the membrane was incubated with avidin-conjugated alkaline phosphatase for 30 min. Proteins were visualized using 5-bromo-4-chloro-3-indolylphosphate/nitroblue tetrazolium (BCIP/NBT) phosphatase substrate (Kirkegaard & Perry Laboratories, Gaithersburg, MD, USA).
Primary antibodies included the general monoclonal anti-αII-spectrin (Affinity Research Products Ltd., Plymouth Meeting, PA; 1:5,000), activated caspase-3 (Cell Signaling, Danvers, MA; 1:1,000) and the cleavage specific antibodies produced by our laboratory including polyclonal anti-SBDP150N (1:1,000), polyclonal anti-SBDP150 (1:1,000) [39] and anti-SBDP145 (1:300) for calpains, polyclonal anti-SBDP150i (1:1,000) and anti-SBDP120 (1:1,000) for caspases and activated calpain-1 (1:300).
Pharmacologic intervention
In addition to untreated controls, the following conditions were used: maitotoxin (MTX) (1 nM; WAKO Chemical USA Inc., Richmond, VA) as a calpain-dominated challenge for 1 or 3 hours; the Ca2+ chelator ethylene diamine tetra-acetic acid (EDTA) (5 mM; Sigma–Aldrich, St. Louis, MO) for 24 h as a caspase-dominated challenge; and staurosporine (STS) (1 μM; Sigma–Aldrich) for 24 h as an excitotoxin challenge [40–42]. For pharmacologic intervention, cultures were pretreated 1 h before the MTX, EDTA or STS challenge with the calpain inhibitor SJA6017 (30 μM; Senju Pharmaceuticals, Kobe, Japan) [43, 44], or the pan-caspase inhibitor Z-VAD (OMe)-FMK (30 μM; R&D, Minneapolis, MN).
Immunocytochemistry
PC-12 cells were seeded onto LabTek II chamber slides (Nunc, Naperville, IL) followed by overnight incubation [45]. The next day, the medium was replaced and treated with MTX, STS or EDTA. Twenty-four hours following STS and EDTA treatment and 3 h for MTX, PC-12 cells were fixed with 4% paraformaldehyde in PBS for 10 min, washed with PBS and permeabilized with 0.1% Triton X-100 in PBS for 5 min. The cells were incubated overnight in blocking solution at 4°C with anti-SBDP145 (1:300), anti-SBDP120 (1:1,000), anti-activated calpain-1 (1:300) or anti-activated caspase-3 (1:1,000) following a 1-h blocking step in 10% goat serum in TBST at room temperature. Alexa 488-conjugated goat-anti-rabbit or mouse secondary antibody (Molecular Probes, Eugene, OR) was added at a dilution of 1:1,000, followed by washing with PBS. The cells were mounted using medium with 4,6-diamidine-2-phenylindole (DAPI) (Vector Laboratories, Burlingame, CA). Fluorescence images were viewed and digitally captured with either a 20× or 40× objective with an Axiovert 200 microscope (Zeiss, Thornwood, NY, USA) equipped with a Spot Real Time Slider high resolution color CCD digital camera (Diagnostic Instruments, Inc., Sterling Heights, MI, USA).
Results
Design of αII-spectrin fragmental-specific antibodies
Analysis of immunoblots and immunocytochemistry for complete and fragmented peptides of non-erythroid αII-spectrin requires antibodies to specific cleavage sites of this protein which we have reported in previous studies and our new N-terminal microsequence [7, 39, 46, 47]. In Fig. 1 the cleavage sites, as highlighted on the partial sequence of αII-spectrin, indicate where the epitopes for the antibodies to these specific αII-SBDPs have been found. SBDP150 (Y*GMMPR) and SBDP145 (G*SAHEV) are calpain (Calp)-cleavage specific fragments while SBDP150i (D*SKTAS) and SBDP120 (D*SVEAL) are caspase (Casp)-cleavage specific. Both proteases cleave αII-spectrin in repeat 11 near the calmodulin-binding domain (CaM) to produce 150 kDa SBDPs with a unique N-terminal region (SBDP150 N). Included in Fig. 1 is an example of an immunoblot with in vitro calpain-2 and caspase-3 digestion of rat brain lysates when probed with αII-spectrin (Biomol, Affinity, USA). As indicated, calpain-2 treatment generated αII-spectrin fragments 150 (SBDP150) and 145 kDa (SBDP145) and caspase-3 generates major fragments of 150 and 120 (SBDP 120). However, when compared to calpain-mediated SBDP150, caspase-3 generated SBDP150 (SBDP150i) appears to have a relatively lower molecular weight around 149 kDa.

Cleavage sites of αII-spectrin. The partial amino acid sequence of αII-spectrin pictured here highlights the cleavage sites of calpains and caspases and demonstrates the approximate sizes of the various SBDPs
Morphology following treatments
PC12 cells treated with either STS, MTX, or EDTA demonstrate the characteristic morphological changes of programmed cell death that accompanies the various treatments. While the control cells maintained the well-defined oval nuclear morphology with diffused DNA of a healthy cell, for the STS and EDTA treated cells the DNA condensation was accompanied by the collapse and shrinkage of the nucleus and eventual disintegration into membrane bound vesicles (Fig. 2). In the MTX-treated cells, a more nonspecific disintegration of the cells and nuclei with their DNA were observed (Fig. 2).

PC12 cell morphology after chemical treatment. PC12 cells were treated with MTX (1 nm), STS (1 mM), and EDTA (5 mM). The cells were observed under phase contrast with DAPI stain, for detecting nuclear morphology, after the noted times. The arrows highlight the typical structure of cells, except control (CTRL) cells, undergoing cell death following treatment. Magnification = 200×, scale bar = 20 mm
AlphaII-spectrin biomarker analysis of treatments
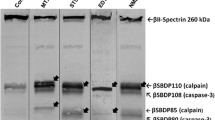
The breakdown products of the protein αII-spectrin (SBDPs) are known biomarkers and are able to distinguish, indirectly, between calpain and caspase activation [7, 47, 48]. The results show that treatment for 1 h with MTX, a Ca2+ influx stimulator, was able to induce the calpain-specific cleavage of αII-spectrin in the PC12 cells (Fig. 3). The antibodies specific to the SBDP150, SBDP150 N and SBDP145, all calpain-cleaved fragments, highlighted increased levels of the fragments over control strongly suggesting calpain activation. Contrast to this is the lack of detectable SBDP150i and SBDP120 fragments indicative of caspase activation such as is seen with the calcium chelator EDTA treated (24 h) PC12 cells (Fig. 3).

αII-SBDPs after treatments. PC12 (rat) neuronal-like cells were treated with MTX (1 nm—1 h), STS (1 mM—24 h), and EDTA (5 mM—24 h). The cell lysates (20 mg) were separated by 6% SDS–PAGE gels. The breakdown products were probed with a general antibody to spectrin (top blot) and with specific antibodies to the breakdown products particular to each family of proteases, SBDP150, SBDP150N and SBDP145 for calpains and SBDP150i and SBDP120 for caspases. All the experiments are done five times independently
For STS treatment, the results after 24 h revealed that both calpain and caspases were activated. STS was generally considered as a non-selective protein kinase inhibitor to function as an inducer of apoptosis. Here it was evidenced by the generation of the caspase-cleaved SBDP150i and SBDP120 fragment. Additionally, STS treatment also generates the calpain-cleaved fragments as the increased levels of SBDP145 demonstrate. Interestingly, no SBDP150 was detected except SBDP150N (Fig. 3).
Correlate SBDPs to the activation of calpain-1 and caspase-3
To more directly assess the activation of calpains and caspases, PC12 cells were treated with MTX, STS and EDTA, with or without 1 h of pretreatment with either the pan-calpain inhibitor SJA6017 (SJA) or the pan-caspase inhibitor Z-VAD-FMK (VAD). In agreement with the αII-spectrin biomarker analysis, the immunoblot results illustrated that calpain-1 was activated following MTX and STS but not by EDTA, while caspase-3, the main caspase executioner, was activated by STS and EDTA treatment but not MTX (Fig. 4). The cells pretreated with the calpain inhibitor SJA showed a visible decrease in the levels of calpain-1 for the MTX-treated cells, an apparent decrease in the STS-treated cells, and it had no effect on the activated caspase-3. On the other hand, the caspase inhibitor VAD was effective in reducing (STS) or nearly eliminating (EDTA) the activation of the caspase-3 by reducing the levels of the 17 kDa, or large subunit, of the active caspase-3 molecule required for an active caspase-3. We further probed the multiple SBDPs and correlated them to the activity of calpain and caspase with either the specific calpain inhibitor SJA6017 or the pan-caspase inhibitor Z-VAD prior to chemical treatments. In Fig. 5, we found the production of SBDP145 and SBDP120 strikingly paralleled the activity of calpain and caspase-3. Calpain or caspase inhibitor successfully blocked SBDP150, SBDP145 and SBDP120 formation. However, it did not block SBDP150N. Even though calpain activation was detected in STS treatment, no SBDP150 could be found in the blots.

Activation of calpain-1 and caspase-3 after treatments. PC12 neuronal-like cells were treated with MTX (M; 1 nm—1 h), STS (S; 1 mM—24 h), and EDTA (E; 5 mM—24 h) in the absence and presence of 1 h pretreatment with either the calpain inhibitor SJA6017 (SJA; 30 mM) or the pan caspase inhibitor Z-VAD-FMK (VAD; 30 mM).The cell lysates (20 mg) were separated by 4–20% gradient SDS–PAGE gels. All the experiments are done three times independently

Effects of calpain and caspase inhibitors on specific SBDP generation pattern. The treated PC12 cell lysates were probed with αII-spectrin (top blot) and specific SBDP-specific antibodies. Paralleled αII-spectrin (top) proteolysis and multiple SBDP were monitored as the marker of calpain or caspase activity. Calpain inhibition preserved the SBDP150 and SBDP145, while caspase inhibition preserved SBDP120. All the gels used in this figure are 6% SDS–PAGE gels except SBDP150i (4–20%). All the experiments are done three times independently
Our results also showed that the antibodies specific to the SBDP150i and SBDP120, both caspase-cleaved fragments in STS and EDTA treatment groups, exhibited increased levels over control and MTX treatment (Fig. 5). However, compared to the STS treatment, EDTA treatment generated less SBDP150i, and caspase-3 inhibitor efficiently prevent SBDP120 formation, but not SBDP150i. More interesting, we also noticed that one new band appeared below SBDP120 in the STS and EDTA treatment panels. This band can be recognized by both total αII-spectrin and the breakdown product specific antibody SBDP120. And, it has the same behavior as SBDP120 which responds to caspase-3 activation.
Immunocytochemical confirmation of increases in active proteosome and SBDPs following treatment
To confirm that activated calpain-1 and its αII-spectrin target fragment, SBDP145, was upregulated following MTX and STS treatments and that increased levels were analogous to the results noted previously using immunoblots (Figs. 4 and 5, respectively) we examined PC12 cells following treatment using immunocytochemistry (Fig. 6). There is a clear upregulation when compared to controls of SBDP145 and calpain-1 in MTX and STS-treated cells but not in the EDTA treated cells which showed similar levels of upregulation to that of the controls.

Activated calpain-1 and its target SBDP145 in PC12 cells challenged with MTX, STS, or EDTA. The PC12 cells were treated with MTX (1 nm—3 h), STS (1 mM—24 h), and EDTA (5 mM—24 h) and then probed with the indicated antibody. Calpain-1 is activated under MTX and STS treatments, but not by EDTA treatment. This is supported by the increased presence of SBDP145. Magnification = 400×, scale bar = 20 mm
Likewise, to confirm that activated caspase-3 and its αII-spectrin target fragment, SBDP120, were upregulated following STS and EDTA treatments and that they, also, were analogous to the results noted previously using immunoblots (Figs. 3 and 4, respectively) we examined PC12 cells following treatment using immunocytochemistry (Fig. 7). There is a clear up-regulation of SBDP120 and caspase-3 in STS and EDTA treated cells, but only a barely detectable up-regulation of caspase-3 in the MTX treated cells and no increased levels in the controls.

Activated caspase-3 and its target SBDP120 in PC12 cells challenged with MTX, STS, or EDTA. The PC12 cells were treated with MTX (1 nm—3 h), STS (1 mM—24 h), and EDTA (5 mM—24 h) and then probed with the indicated antibody. Caspase-3 is activated under STS and EDTA treatments and this is supported by the increased presence of SBDP120, but little active caspase-3 or its target SBDP120 is seen in the MTX-treated cells. Magnification = 400×, scale bar = 20 mm
Discussion
This is the first systematic study to characterize αII-SBDPs in different cell death models with fragment-specific SBDP antibodies. In this study, three distinct chemical challenged cell death models were used to induce calpain-dominant necrosis, caspase-dominant apoptosis and an active mixture of the two (Fig. 8).

Schematic of pathways leading to cell death when cells are challenged with MTX, STS, or EDTA. The schematic represents how the different treatments lead to different forms of cell death, oncosis (necrosis) or apoptosis. The processing of the biomarker αII-spectrin under the various treatments indicates which protease, calpain or caspase, is activated under the different paradigms
MTX, a highly potent marine toxin isolated from the dinoflagellate gambierdiscus toxicus, is an important molecular tool for the study of oncotic (necrotic) cell death [49, 50]. MTX, known for its ability to stimulate calcium influx into cells through a series of biochemical processes that include activation of both voltage-sensitive and receptor-operated calcium channels [51, 52], initiates calpain activation which eventually leads to cell death via cell lysis [40]. In our study, we confirmed that total αII-spectrin was degraded into SBDP150 and SBDP145 by calpain-dominant MTX-induced necrosis.
Staurosporine is an indolo[2,3-a]carbazole discovered in the course of screening extracts of the bacterium Streptomyces staurospores for constituent alkaloids with PKC-inhibitory activity. STS was shown to mobilize and increase [Ca2+] from the thapsigargin-insensitive secretory granule Ca2+ stores by inhibiting protein kinase C [53]. Now identified as a potent, relatively non-selective, protein kinase inhibitor, recognized to block, by varying degrees, a number of different kinases, it is commonly employed as an intracellular stress inducer of apoptosis [54, 55]. However, it is controversial in that it not only induces caspase-dominant activation, but also calpain activation [56–58]. The precise mechanism underlying its activity is not known yet. In this study, our fragment-specific antibodies recognized SBDP150N, SBDP150i, SBDP145 and SBDP120 in STS-treated PC12 cells. Most interestingly SBDP150 could not be detected in STS challenged PC12 cells. In addition, although the calpain inhibitor did not prevent SBDP150N formation, it was able to prevent SBDP145. These results suggested that STS induces both calpain and caspase activation. STS appears to alter Ca2+ homeostasis of differentiated PC12 cells. Seo and Seo [59] found that the anti-apoptotic Bcl-2 family member Bcl-XL prevented STS-induced neuronal cell death by interfering with Ca2+-mediated apoptotic signals by inhibiting Ca2+ release from ER. This is consistent with the results obtained by Wang and colleagues [60] in rat hepatocytes who found that Bcl-XL prevented cytochrome c release and caspase-3 activation in response to STS. It was also reported that less Ca2+ was released from the ER in Bcl-XL expressing cells in response to apoptotic stimuli due to down-regulation of IP3 receptors [61]. Moreover, a proteomic study by Short et al. [55] indicated that STS-induced apoptosis induces an unfolded protein response involving molecular chaperones, co-chaperones and structural proteins indicative of ER stress. On the other hand Kim et al. [53] report that they did not find the ER to be the source of increased cytosolic Ca2+ concentration but instead they suggested secretory granules in the rat submandibular acinar cells to be the responsive elements to STS treatment [53]. Whether STS increases the cytosolic Ca2+ concentration from the ER Ca2+ stores, secretory granules or from external sources, Ca2+ appeared to play an important role in activating calpain and generating the calpain-cleaved fragments as the increased levels of SBDP145 demonstrated. This is not to overlook or minimize the crosstalk that may occur such as calpain contributing to caspase activation [58]. It has been reported that calpain-2 also cleaves caspase-3 and renders it more susceptible to full activation after neonatal hypoxia–ischemia [7, 62].
Based on our results, we propose that αII-spectrin first generates SBDP150N, SBDP150 and then SBDP145 under calpain-dominant STS challenge. Caspase-3 activation on the other hand cleaves SBDP150 to generate SBDP150i and then SBDP120. These results suggest SBDP145 is more sensitive and specific for calpain activation when compared to SBDP150 and SBDP150N. Conceptually we hypothesized that the SBDP150N antibody would behave similar to the SBDP150 antibody when we designed the peptides for it. Although our results suggest that SBDP150N generates the same patterns in calpain-dominant treatment (MTX) with or without calpain inhibition as SBDP150 and SBDP145; it, however, behaves differently in mixed calpain and caspase activated conditions (STS). It appears that calpain inhibition (SJA) is not able to prevent SBDP150N formation. The exact reaction underlying this phenomenon is unknown yet. We think that STS treatment generated a caspase dominant SBDP150i when the calpain inhibitor was added. Since there is only nine amino acids span difference between SBDP150N and SBDP150i, and based upon the native quaternary structure, it is possible that SBDP150N antibody picked up not only SBDP150N, but also some SBDP150i fragments under the calpain inhibition condition.
EDTA, a chemical compound also known as diaminoethanetetraacetic acid disodium salt, edetate, or versene, is a chelating agent. It forms coordination compounds with most monovalent, divalent, trivalent and tetravalent metal ions, such as silver (Ag+), calcium (Ca2+), Zinc (Zn2+), copper (Cu2+), iron (Fe3+) or zirconium (Zr4+). Ethylene glycol tetra-acetic acid (EGTA), a chelating agent related to EDTA, also has a high affinity for calcium and was previously found to trigger apoptosis in SH-SY5Y [41] and L929 mouse skin fibroblast cells [63]. Since EDTA is a calcium chelator and as such removes most if not all of the calcium in cells it triggers a stress response by the endoplasmic reticulum (ER) known as the unfolded protein response (UPR). The response is due to the disruption of cellular Ca2+ homeostasis disrupting the critical role Ca2+ plays as a co-factor in the synthesis and the proper folding of proteins, within the ER lumen [41, 64]. It has been also reported that EDTA induces apoptosis by Zn2+ chelation in Neuro-2A cell lines [65] or disrupting calcium-dependent cell adhesion molecules on intestinal epithelial cells [66]. However, those exact mechanisms are still under investigation.
Our results showed EDTA triggering a typical apoptosis like response in neuronal-like PC12 cells with caspase-3 activation. Typical of this response αII-spectrin was degraded to SBDP150i and SBDP120 after caspase-3 was activated. Consistent with our previous N-terminal micro-sequence study, this confirms our finding that calpain-mediated SBDP150 is different from caspase-3 mediated SBDP150i due to a nine residue span by fragment-specific antibodies.
Our results showed that SBDP150i and SBDP120 exhibited increased levels upon STS or EDTA treatment. The level of SBDP150i is less in EDTA treatment compared to STS groups. This might be due to more of it being converted from SBDP150i to SBDP120. However, compared to SBDP120, caspase inhibitor z-VAD does not efficiently block SBDP150i formation. This might due to several reasons: first, since SBDP150i is the first caspase cleavage site, it will be more susceptible and harder to block compared to SBDP120. Second, it has been reported that caspase-3 is not the only caspase involved. Other caspases including caspase-1, 2 4, 6 and 7 may also be involved in SBDP150i formation [47, 67]. However, SBDP120 is generated only by caspase-3. Caspase inhibitor Z-VAD at 30 μM appears most effective against caspase-3 and not as much as against the other caspases.
Another interesting phenomenon we observed is that an additional low molecular weight SBDP120 band appears upon ex vivo STS and EDTA treatment and it responds to the caspase inhibitor Z-VAD. The induction of the doublet of SBDP120 has also been occasionally noticed in the STS and EDTA treated human SY5Y and rat primary cortical neuronal cells (data not shown). However, we could not detect the doublet SBDP120 in any in vitro caspase-3 digested samples. Moreover, not all STS and EDTA treated cell lines can generate this doublet SBDP120. Based on the fact that our caspase-3 cleavage specific (D*SVEAL) antibody SBDP120 can detected both SBDP120 bands, we therefore assume that the generation of the lower molecular weight SBDP120 is a dynamic event. After active caspase-3 cleaves αII-spectrin to SBDP120, then caspase-3 or another apoptotic related proteases may further cleave spectrin at the C-terminal at some critical time point creating the slightly lower molecular weight SBDP120 (lower band of SBDP120). The exact mechanism underlying the SBDP120 generation needs to be further clarified.
In addition to PC12 cells, we found that EDTA was also a better than average caspase-3 activator in a number of other cell types, including rat cerebrocortical and cerebellar granule neurons (data not shown). This suggests EDTA challenged neurons should provide a good model for studying caspase-3 related neuronal diseases.
Taken together, our studies have demonstrated that the multiple αII-SBDPs can be biomarkers capable of distinguishing calpain and caspase dominant necrotic and apoptotic cell death and is able to monitor the dynamic biochemical changes associated with calpain and caspase activation. Furthermore, the antibodies developed here, such as calpain specific SBDP145 and caspase-3 specific SBDP120 could be invaluable tools to monitor disease progression and the consequences of inhibitory potency of drugs that target calpain and caspase activation in different neuronal disease models such as ischemic brain injury, traumatic brain injury and Alzheimer’s disease.
Abbreviations
- SBDP:
-
Spectrin breakdown product
- MTX:
-
Maitotoxin
- STS:
-
Staurosporine
- EDTA:
-
Ethylenediaminetetraacetic acid
- TBI:
-
Traumatic brain injury
- PCD:
-
Programmed cell death
References
Raghupathi R, Graham DI, McIntosh TK (2000) Apoptosis after traumatic brain injury. J Neurotrauma 17:927–938
Pike BR, Flint J, Dave JR et al (2004) Accumulation of calpain and caspase-3 proteolytic fragments of brain-derived alphaII-spectrin in cerebral spinal fluid after middle cerebral artery occlusion in rats. J Cereb Blood Flow Metab 24:98–106
Behl C (2000) Apoptosis and Alzheimer’s disease. J Neural Transm 107:1325–1344
Clark RS, Chen J, Watkins SC et al (1997) Apoptosis-suppressor gene bcl-2 expression after traumatic brain injury in rats. J Neurosci 17:9172–9182
Reed JC (2000) Mechanisms of apoptosis. Am J Pathol 157:1415–1430
Raghupathi R (2004) Cell death mechanisms following traumatic brain injury. Brain Pathol 14:215–222
Wang KK (2000) Calpain and caspase: can you tell the difference? Trends Neurosci 23:20–26
Beer R, Franz G, Srinivasan A et al (2000) Temporal profile and cell subtype distribution of activated caspase-3 following experimental traumatic brain injury. J Neurochem 75:1264–1273
Vanderklish PW, Bahr BA (2000) The pathogenic activation of calpain: a marker and mediator of cellular toxicity and disease states. Int J Exp Pathol 81:323–339
Majno G, Joris I (1995) Apoptosis, oncosis, and necrosis. An overview of cell death. Am J Pathol 146:3–15
Van Cruchten S, Van Den Broeck W (2002) Morphological and biochemical aspects of apoptosis, oncosis and necrosis. Anat Histol Embryol 31:214–223
Lecoeur H, Prevost MC, Gougeon ML (2001) Oncosis is associated with exposure of phosphatidylserine residues on the outside layer of the plasma membrane: a reconsideration of the specificity of the annexin V/propidium iodide assay. Cytometry 44:65–72
Rathmell JC, Thompson CB (1999) The central effectors of cell death in the immune system. Annu Rev Immunol 17:781–828
Lockshin RA, Zakeri Z (2004) Caspase-independent cell death? Oncogene 23:2766–2773
Susin SA, Zamzami N, Castedo M et al (1997) The central executioner of apoptosis: multiple connections between protease activation and mitochondria in Fas/APO-1/CD95- and ceramide-induced apoptosis. J Exp Med 186:25–37
Kerr JF, Wyllie AH, Currie AR (1972) Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer 26:239–257
Castillo MR, Babson JR (1998) Ca(2+)-dependent mechanisms of cell injury in cultured cortical neurons. Neuroscience 86:1133–1144
Farber E (1994) Programmed cell death: necrosis versus apoptosis. Mod Pathol 7:605–609
Levin S (1998) Apoptosis, necrosis, or oncosis: what is your diagnosis? A report from the Cell Death Nomenclature Committee of the Society of Toxicologic Pathologists. Toxicol Sci 41:155–156
Hirsch T, Marchetti P, Susin SA et al (1997) The apoptosis-necrosis paradox. Apoptogenic proteases activated after mitochondrial permeability transition determine the mode of cell death. Oncogene 15:1573–1581
Criddle DN, Gerasimenko JV, Baumgartner HK et al (2007) Calcium signalling and pancreatic cell death: apoptosis or necrosis? Cell Death Differ 14:1285–1294
Muller GJ, Stadelmann C, Bastholm L, Elling F, Lassmann H, Johansen FF (2004) Ischemia leads to apoptosis- and necrosis-like neuron death in the ischemic rat hippocampus. Brain Pathol 14:415–424
Friedman LM, Furberg CD, DeMets DL (1998) Fundamentals of clinical trials, 3rd edn. Springer-Verlag, St Louis, MO
Lewis SB, Velat GJ, Miralia L et al (2007) Alpha-II spectrin breakdown products in aneurysmal subarachnoid hemorrhage: a novel biomarker of proteolytic injury. J Neurosurg 107:792–796
Ringger NC, O’Steen BE, Brabham JG et al (2004) A novel marker for traumatic brain injury: CSF alphaII-spectrin breakdown product levels. J Neurotrauma 21:1443–1456
Pike BR, Flint J, Dutta S, Johnson E, Wang KK, Hayes RL (2001) Accumulation of non-erythroid alpha II-spectrin and calpain-cleaved alpha II-spectrin breakdown products in cerebrospinal fluid after traumatic brain injury in rats. J Neurochem 78:1297–1306
Cardali S, Maugeri R (2006) Detection of alphaII-spectrin and breakdown products in humans after severe traumatic brain injury. J Neurosurg Sci 50:25–31
Huh GY, Glantz SB, Je S, Morrow JS, Kim JH (2001) Calpain proteolysis of alpha II-spectrin in the normal adult human brain. Neurosci Lett 316:41–44
Riederer BM, Zagon IS, Goodman SR (1986) Brain spectrin(240/235) and brain spectrin(240/235E): two distinct spectrin subtypes with different locations within mammalian neural cells. J Cell Biol 102:2088–2097
Meary F, Metral S, Ferreira C et al (2007) A mutant alphaII-spectrin designed to resist calpain and caspase cleavage questions the functional importance of this process in vivo. J Biol Chem 282:14226–14237
Povlishock JT (1992) Traumatically induced axonal injury: pathogenesis and pathobiological implications. Brain Pathol 2:1–12
Povlishock JT, Stone JR (2001) Traumatic axonal injury. In: Langston JW (ed) Head trauma basic, preclinical, and clinical directions. Wiley, New York, pp. 281–301
Bouvry D, Planes C, Malbert-Colas L, Escabasse V, Clerici C (2006) Hypoxia-induced cytoskeleton disruption in alveolar epithelial cells. Am J Respir Cell Mol Biol 35:519–527
Das A, Belagodu A, Reiter RJ, Ray SK, Banik NL (2008) Cytoprotective effects of melatonin on C6 astroglial cells exposed to glutamate excitotoxicity and oxidative stress. J Pineal Res 45:117–124
Del Rio P, Montiel T, Massieu L (2008) Contribution of NMDA and non-NMDA receptors to in vivo glutamate-induced calpain activation in the rat striatum. Relation to neuronal damage. Neurochem Res 33:1475–1483
Kawamura M, Nakajima W, Ishida A, Ohmura A, Miura S, Takada G (2005) Calpain inhibitor MDL 28170 protects hypoxic-ischemic brain injury in neonatal rats by inhibition of both apoptosis and necrosis. Brain Res 1037:59–69
Koumura A, Nonaka Y, Hyakkoku K et al (2008) A novel calpain inhibitor, ((1S)-1((((1S)-1-benzyl-3-cyclopropylamino-2, 3-di-oxopropyl)amino)carbonyl)-3-methylbutyl) carbamic acid 5-methoxy-3-oxapentyl ester, protects neuronal cells from cerebral ischemia-induced damage in mice. Neuroscience 157:309–318
Tamada Y, Nakajima E, Nakajima T, Shearer TR, Azuma M (2005) Proteolysis of neuronal cytoskeletal proteins by calpain contributes to rat retinal cell death induced by hypoxia. Brain Res 1050:148–155
Dutta S, Chiu YC, Probert AW, Wang KK (2002) Selective release of calpain produced alphalI-spectrin (alpha-fodrin) breakdown products by acute neuronal cell death. Biol Chem 383:785–791
Wang KK, Nath R, Raser KJ, Hajimohammadreza I (1996) Maitotoxin induces calpain activation in SH-SY5Y neuroblastoma cells and cerebrocortical cultures. Arch Biochem Biophys 331:208–214
McGinnis KM, Wang KK, Gnegy ME (1999) Alterations of extracellular calcium elicit selective modes of cell death and protease activation in SH-SY5Y human neuroblastoma cells. J Neurochem 72:1853–1863
Nath R, Scott M, Nadimpalli R, Gupta R, Wang KK (2000) Activation of apoptosis-linked caspase(s) in NMDA-injured brains in neonatal rats. Neurochem Int 36:119–126
Fukiage C, Azuma M, Nakamura Y, Tamada Y, Nakamura M, Shearer TR (1997) SJA6017, a newly synthesized peptide aldehyde inhibitor of calpain: amelioration of cataract in cultured rat lenses. Biochim Biophys Acta 1361:304–312
Kupina NC, Nath R, Bernath EE et al (2001) The novel calpain inhibitor SJA6017 improves functional outcome after delayed administration in a mouse model of diffuse brain injury. J Neurotrauma 18:1229–1240
Zhang Z, Ottens AK, Larner SF et al (2006) Direct Rho-associated kinase inhibiton induces cofilin dephosphorylation and neurite outgrowth in PC-12 cells. Cell Mol Biol Lett 11:12–29
Nath R, Davis M, Probert AW et al (2000) Processing of cdk5 activator p35 to its truncated form (p25) by calpain in acutely injured neuronal cells. Biochem Biophys Res Commun 274:16–21
Wang KK, Posmantur R, Nath R et al (1998) Simultaneous degradation of alphaII- and betaII-spectrin by caspase 3 (CPP32) in apoptotic cells. J Biol Chem 273:22490–22497
Pike BR, Zhao X, Newcomb JK, Posmantur RM, Wang KK, Hayes RL (1998) Regional calpain and caspase-3 proteolysis of alpha-spectrin after traumatic brain injury. Neuroreport 9:2437–2442
Schilling WP, Sinkins WG, Estacion M (1999) Maitotoxin activates a nonselective cation channel and a P2Z/P2X(7)-like cytolytic pore in human skin fibroblasts. Am J Physiol 277:C755–C765
Schilling WP, Wasylyna T, Dubyak GR, Humphreys BD, Sinkins WG (1999) Maitotoxin and P2Z/P2X(7) purinergic receptor stimulation activate a common cytolytic pore. Am J Physiol 277:C766–C776
Meucci O, Grimaldi M, Scorziello A et al (1992) Maitotoxin-induced intracellular calcium rise in PC12 cells: involvement of dihydropyridine-sensitive and omega-conotoxin-sensitive calcium channels and phosphoinositide breakdown. J Neurochem 59:679–688
Soergel DG, Yasumoto T, Daly JW, Gusovsky F (1992) Maitotoxin effects are blocked by SK&F 96365, an inhibitor of receptor-mediated calcium entry. Mol Pharmacol 41:487–493
Kim YJ, An JM, Shin DM, Lee SI, Sugiya H, Seo JT (2002) Staurosporine mobilizes Ca(2+) from secretory granules by inhibiting protein kinase C in rat submandibular acinar cells. J Dent Res 81:788–793
Koh JY, Wie MB, Gwag BJ et al (1995) Staurosporine-induced neuronal apoptosis. Exp Neurol 135:153–159
Short DM, Heron ID, Birse-Archbold JL, Kerr LE, Sharkey J, McCulloch J (2007) Apoptosis induced by staurosporine alters chaperone and endoplasmic reticulum proteins: Identification by quantitative proteomics. Proteomics 7:3085–3096
Deshmukh M, Johnson EM Jr (2000) Staurosporine-induced neuronal death: multiple mechanisms and methodological implications. Cell Death Differ 7:250–261
Zhang XD, Gillespie SK, Hersey P (2004) Staurosporine induces apoptosis of melanoma by both caspase-dependent and -independent apoptotic pathways. Mol Cancer Ther 3:187–197
Neumar RW, Xu YA, Gada H, Guttmann RP, Siman R (2003) Cross-talk between calpain and caspase proteolytic systems during neuronal apoptosis. J Biol Chem 278:14162–14167
Seo SR, Seo JT (2009) Calcium overload is essential for the acceleration of staurosporine-induced cell death following neuronal differentiation in PC12 cells. Exp Mol Med 41:269–276
Wang Y, Zhang B, Peng X, Perpetua M, Harbrecht BG (2008) Bcl-xL prevents staurosporine-induced hepatocyte apoptosis by restoring protein kinase B/mitogen-activated protein kinase activity and mitochondria integrity. J Cell Physiol 215:676–683
Li C, Fox CJ, Master SR, Bindokas VP, Chodosh LA, Thompson CB (2002) Bcl-X(L) affects Ca(2+) homeostasis by altering expression of inositol 1,4,5-trisphosphate receptors. Proc Natl Acad Sci USA 99:9830–9835
Blomgren K, Zhu C, Wang X et al (2001) Synergistic activation of caspase-3 by m-calpain after neonatal hypoxia–ischemia: a mechanism of “pathological apoptosis”? J Biol Chem 276:10191–10198
Serper A, Calt S, Dogan AL, Guc D, Ozcelik B, Kuraner T (2001) Comparison of the cytotoxic effects and smear layer removing capacity of oxidative potential water, NaOCl and EDTA. J Oral Sci 43:233–238
Chiesa R, Angeretti N, Del Bo R, Lucca E, Munna E, Forloni G (1998) Extracellular calcium deprivation in astrocytes: regulation of mRNA expression and apoptosis. J Neurochem 70:1474–1483
Sakabe I, Paul S, Dansithong W, Shinozawa T (1998) Induction of apoptosis in Neuro-2A cells by Zn2+ chelating. Cell Struct Funct 23:95–99
Abud HE, Heath JK (2004) Detecting apoptosis during the formation of polarized intestinal epithelium in organ culture. Cell Death Differ 11:788–789
Rotter B, Kroviarski Y, Nicolas G, Dhermy D, Lecomte MC (2004) AlphaII-spectrin is an in vitro target for caspase-2, and its cleavage is regulated by calmodulin binding. Biochem J 378:161–168
Acknowledgements
We wish to thank Meghan O’Donoghue for her technical assistance. This work was supported by the Department of Defense grant DAMMED-03-1-0066, National Institutes of Health grants R01 NS049175-01-A1 and R01 NS051431. K.K.W. Wang and R.L. Hayes own stock of Banyan Biomarkers Inc., and may benefit financially as a result of the outcome of this research or the work reported in this publication.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Zhiqun Zhang and Stephen F. Larner contributed equally to this work.
Rights and permissions
About this article
Cite this article
Zhang, Z., Larner, S.F., Liu, M.C. et al. Multiple alphaII-spectrin breakdown products distinguish calpain and caspase dominated necrotic and apoptotic cell death pathways. Apoptosis 14, 1289–1298 (2009). https://doi.org/10.1007/s10495-009-0405-z
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10495-009-0405-z