
Abstract
Ardipusilloside III is a saponin newly isolated from Ardisia pusilla A.DC. Since saponins have exhibited broad anti-cancer and pro-apoptotic activity, we investigated the ability of ardipusilloside III to induce apoptosis in human glioblastoma U251MG cells, as well as the involvement of apoptotic signaling pathways. Ardipusilloside III markedly suppressed proliferation of U251MG cells in a time- and dose-dependent manner (P < 0.05, IC50 = 8.2 μg/ml), but did not affect the growth of primary cultures of human astrocytes. Ardipusilloside III-treated U251MG cells underwent typical apoptotic changes. Exposure to a low dose of ardipusilloside III provoked G2/M-phase cell cycle arrest, which preceded apoptosis characterized by the appearance of cells with sub-G1 DNA content. However, a higher dose of ardipusilloside III induced apoptosis without first causing cell cycle arrest. In addition, ardipusilloside III exposure resulted in time-dependent BAD dephosphorylation and cleavage as well as activation of caspase-8 and caspase-3. Therefore, both the intrinsic pathway of apoptosis, mediated by BAD dephosphorylation and cleavage, and the extrinisic pathway of apoptosis, mediated by caspase-8 and caspase-3 activation, were involved in ardipusilloside III-induced apoptosis. These data suggest that ardipusilloside III is a reliable candidate for chemotherapeutic treatment of human glioblastomas, and should be investigated further.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Glioblastoma multiforme (WHO Grade IV), composed of poorly differentiated neoplastic astrocytes, is the most common and lethal primary brain malignancy [1]. Despite the combination of treatments offered to patients, including selective surgery, radiotherapy, and chemotherapy, residual cancer cells inevitably invade the surrounding normal brain tissue, leading to cancer recurrence and a poor prognosis. Since glioblastomas are relatively resistant to radiation and chemotherapy due to genetic and epigenetic alterations, development of effective drugs to reverse its drug resistance and induce apoptosis is critical [2, 3].
The discovery of novel anti-cancer agents has progressed considerably in recent years. Compelling data from laboratory studies, epidemiological investigations, and human clinical trials all have demonstrated that saponins have significant potential in cancer chemoprevention and chemotherapy [4–6]. Saponins also have a neuroprotective effect [7] as well as an immunological adjuvant function [8], which could also provide relief from the development of serious lesions in patients with glioblastoma. However, the mechanisms by which saponins induce apoptosis are not clear.
Tumor growth is the result of either improper cell proliferation or cell death. Necrosis and apoptosis are the two principal mechanisms of cell death. Apoptosis, in contrast to necrosis, does not lead to cell lysis and inflammatory responses in cancer chemotherapy. Avoiding and controlling intense inflammatory responses could prevent fatal encephaledema. Therefore, increasing apoptosis without increasing necrosis during chemotherapeutic treatment of glioblastoma is ideal. The intracellular pathways leading to apoptotic cell death are complicated, but most involve activation of specific proteases known as caspases. There are two primary mechanisms by which caspases are activated: the extrinsic, or death receptor-mediated, pathway and the intrinsic, or mitochondrial-mediated, pathway [9]. The majority of known apoptotic signals are part of the mitochondrial pathway, including both the pro-apoptotic and the anti-apoptotic Bcl-2 family members, loss of mitochondrial transmembrane potential, and release of caspase activators. For death receptor-induced apoptosis, the crucial events are the formation of a Death Inducing Signaling Complex (DISC) and caspase-8 activation [10].
BAD, a member of the Bcl-2 homology 3 (BH3)-only subgroup of the Bcl-2 family as well as a ligand of the prosurvival protein 14-3-3, is a switch protein for the mitochondrial-induced apoptotic pathway [11]. Lacking a C-terminal hydrophobic domain essential for targeting the mitochondrial outer membrane, BAD distributes exclusively in cytoplasm, heterodimerized with 14-3-3. Both BAD dephosphorylation and cytochrome c release are essential to induce apoptosis [12]. In response to cell death stimuli, dephosphorylated BAD translocates to the outer mitochondrial membrane, where it displaces Bax from the Bcl-xL:Bax complex. The newly released Bax then forms a transmembrane pore across the outer mitochondrial membrane, leading to loss of membrane potential and efflux of cytochrome c and the apoptosis-inducing factor (AIF) [9, 13]. BAD also can promote the formation of the mitochondrial permeability transition pore directly [14] and mediate cell cycle progression by regulating the expression of cyclins [15]. During apoptotic cell death, BAD is cleaved by a caspase(s) at its N terminus to a truncated form. The 15-kDa truncated BAD termed by tBADS is a more potent inducer of apoptosis than uncleaved BAD. Previous studies have shown that many anti-cancer agents such as doxorubicin and genistein induce apoptosis through the BAD apoptotic signaling pathway, as well as endogenous signals such as transforming growth factor (TGF)-β1 [16–18]. However, the role of these pathways in saponin-induced apoptosis has not been verified.
Ardisia pusilla A. DC (of the family Myrsinaceae), a Chinese medicinal herb also known as Jiu Jie Long, is the source of the triterpenoid saponins called ardipusillosides. Ardipusilloside I and ardipusilloside II have known anti-cancer activity in both Lewis pulmonary carcinoma and hepatocarcinoma [19, 20]. Recently, the structure of ardipusilloside III, a newly discovered saponin, has been determined by extensive use of nuclear magnetic resonance (NMR) techniques as well as chemical evidence, although this work has not been published yet. Ardipusilloside III is distinguished from ardipusilloside I by the extra 28α-hydroxy group in its aglycone, and different sugar moieties. Since the effect of this new agent on cancer cells has not been tested previously, we investigate herein the ability of ardipusilloside III to induce apoptosis in human glioblastoma U251MG cells. In addition, we seek evidence of the molecular mechanism by which this potential chemotherapeutic agent kills cancer cells.
Materials and methods
Sample preparation
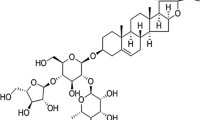
Purified ardipusilloside III (Purity > 98%) was supplied by the Department of Pharmacy of Xijing Hospital, the Fourth Military Medical University, Xi’an, P.R.China. Ardipusilloside III was isolated from A. pusilla and established as 3-O-{β-d-xylopyranosyl-(1→2)-β-d-glucopyranosyl-(1→4)-[β-d-glucopyranosyl-(1→2)]-α-l-arabinopyranosyl}-3β, 16α, 28α-trihydroxy-13β, 28-epoxy-oleanan-30-al with the molecular formula C52H84O23 (molecular weight: 1,076). The structure of this compound is shown in Fig. 1a. Stock solutions were prepared by dissolving the dry material in dimethylsulfoxide (DMSO) (Gibco/Invitrogen, NY, USA) and storing aliquots at −20°C. These were later diluted to the final concentration in fresh medium before each experiment. In all experiments, the final DMSO concentration did not exceed 1‰ (v/v), so as not to affect cell growth. Cells incubated with ardipusilloside III-free medium were used as controls.

In vitro treatment of human glioblastoma cells with adipusilloside III inhibits proliferation in a dose- and time-dependent manner: (a) The chemical structure of adipusilloside III; (b) Dose- and time-dependent effects of adipusilloside III on the viability of human glioblastoma U251MG cells and primary cultured human astrocytes. Columns, mean percent of viable cells from three experiments: Bars, ±SE; *P < 0.05; **P < 0.01, versus control group (ardipusilloside III -free)
Cell lines and cell culture
U251MG human glioblastoma cells (maintained in our laboratory, originally obtained from Uppsala, Sweden) were cultured in DMEM medium supplemented with 10% newborn calf serum (both from Gibco/Invitrogen, NY, USA) in a 37°C incubator with a humidified atmosphere of 5% CO2–95% O2.
Cultured primary astrocytes were obtained from a slightly impaired brain tissue fragment of a volunteer with cerebral trauma who consented to the procedure. Acquisition of the tissue was done following approval by the local medical research ethics committee at Xijing Hospital, the Fourth Military Medical University, Xi’an, P.R. China. The fragment was dissected for its grey matter by a surgeon when it was removed, washed in phosphate buffered sodium (PBS) and dispersed repeatedly. The resulting cell suspension was filtered and cultured in DMEM with 10% newborn calf serum. After 2 weeks in culture, the remaining cells were mostly astrocytes [21].
Twenty-four hours before the experiments, the cells were transferred to serum free medium. Ardipusilloside III of different concentrations was added to the culture medium for different incubation periods as indicated in the Results section.
MTT assay for cytotoxicity
The number of viable cells was determined by the 3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide (MTT) assay as described previously [22]. Briefly, the cells were cultured in 96-well plates (Greiner Bio-One GmbH, Frickenhausen, Germany) at a density of 4 × 103 cells/well in the presence of ardipusilloside III at the indicated concentrations. After incubation for 6, 24, 48, or 72 h, the MTT (Sigma, St. Louis, MO, USA) solved in PBS was added to each well at a final concentration of 5 mg/ml and then incubated at 37°C for 4 h. The water-insoluble dark blue formazan crystals that formed during MTT cleavage in actively metabolizing cells were dissolved in DMSO. The optical density was measured at a wavelength of 490 nm with a Bio-Rad 680 microplate reader (Bio-Rad, California, USA). The reduction in viability of the ardipusilloside III-treated U251MG cells and astrocytes was expressed as the percentage compared with ardipusilloside III-free control cells. All experiments were done in triplicate.
Cell cycle analysis
Cell cycle distribution was analyzed by flow cytometric analysis as described previously [23]. Briefly, following incubating for 12, 24, 48, or 72 h, U251MG cells treated with or without ardipusilloside III were trypsinized with 0.25% trypsin (Sigma, St. Louis, MO, USA), counted, centrifuged at 300g for 5 min, and fixed in ethanol at 4°C overnight. Then the cells were washed and centrifuged. The resulting cell pellets were resuspended in an RNase solution (0.02 mg/ml; Sigma) containing propidium iodide (0.02 mg/ml; Sigma), and incubated at 4°C for 30 min. The DNA content of approximately 1–2 × 105 stained cells were analyzed using a FACScan flow cytometer equipped with the FACStation data management system running Cell Quest software (Becton Dickinson, San Jose, CA, USA). The results are expressed as a plot of fluorescence intensity versus cell number.
Electron microscopy
U251MG cells were cultured in T-150 flasks (Greiner Bio-One GmbH, Frickenhausen, Germany) (3 × 106 cells/cm2) and treated with 8.2 μg/ml ardipusilloside III for 24 h, and then trypsinized with 0.25% trypsin and centrifuged at 1,400g for 15 min. The pellets were fixed in 0.1 M PBS (pH 7.4) with 2.5% glutaraldehyde, then postfixed in 2% buffered osmium tetroxide for 2 h and finally dehydrated through a series of graded ethyl alcohols from 70% to 100%. The schedule is as follows: 70% for 10 min., 95% for 10 min. and three changes of 100% for 5 min each. Specimens used for transmission electron microscopy were embedded in epon resin. Thin sections were cut on an ultramicrotome and double stained with uranyl acetate and lead citrate. Electron micrographs were taken on an electron microscope (JEM-2000EX, JEOL Ltd., Tokyo, Japan) operating at 80 kV.
Annexin V/propidium iodide
To determine a number of apoptotic cells, Annexin V assays were performed using an apoptosis detection kit (Annexin V-FITC/PI Staining Kit; Immunotech Co., Marseille, France). Briefly, 1.5 × 105 cells were plated in 24-well plates, and treated with 3.7 and 8.2 μg/ml ardipusilloside III for 6 and 12 h, respectively. Cells were harvested by treatment with 0.02% EDTA in PBS or by scraping, washed in cold PBS (0.01 mol/l, pH = 7.4), incubated for 15 min with fluorescein-conjugated annexin V and propidium iodide, and analyzed using the same flow cytometer and software used for the cell cycle analysis. PI-negative but annexin V-positive cells were considered to be early apoptotic, while cells that were both PI and annexin V negative were considered normal.
Analysis of DNA fragmentation
Cellular DNA was extracted from the cells according to Gong’s modified method [24]. Briefly, cells treated with or without 8.2 μg/ml ardipusilloside III treatment for 24 h were trypsinized with 0.25% trypsin and collected by centrifugation (200g, 10 min), washed twice in cold PBS (0.01 mol/l, pH = 7.4) and resuspended at a density of 4 × 106 cells/400 μl in hypotonic lysing buffer (5 mM Tris, 20 mM EDTA, pH 7.4) containing 0.5% Triton X-100 for 30 min at 4°C. The lysates were centrifuged at 13,000g for 15 min at 4°C. Fragmented DNA was extracted from the supernatant with phenol-chloroform-isoamylalcohol, precipitated by addition of 2 vol. of absolute ethanol and 0.1 vol. of 3 mM sodium acetate, and treated with RNAse A (500 U/ml) at 37°C for 3 h. The pattern of DNA fragmentation was visualized by electrophoresis in 1.5% agarose gel containing ethidium bromide in 40 mM Tris-acetate buffer (pH 7.5) at 50 V for 4 h and photographed under UV light.
Cellular and nuclear morphology
Apoptotic U251MG cells were identified based on alterations in their nuclear morphology detected by staining with Hoechst 33342 as described previously [25]. Briefly, cells were grown in 24-well plates (1 × 104 cells/well) in the presence of 3.7 and 8.2 μg/ml ardipusilloside III for 24 h. Cells were observed with an inverted microscope (Leica Microsystems, Wetzlar, Germany), then were washed in PBS and fixed in 70% ethanol for 2 h at 4°C. Cell nuclei were stained with 5 μg/ml Hoechst 33342 (Sigma, St. Louis, MO, USA). After final washing in PBS, the changes in nuclear morphology were visualized by fluorescence microscopy (Leica Microsystems, Wetzlar, Germany) using excitation at wavelength of 330–380 nm.
Western blot analysis
Protein extracts of U251MG cells (ardipusilloside III-free control and cells treated with 8.2 μg/ml ardipusilloside III for 6, 12, or 24 h) were prepared by lysing the cells in RIPA buffer (150 mM NaCl, 1% Non-idet P-40, 0.5% sodium deoxycholate, 0.1% SDS, 50 mM Tris-HCl, pH 8) containing 10 mM EDTA, 1 mM sodium orthovanadate and 1 mM phenylmethylsulfonylfluoride (PMSF; all from Sigma) for 30 min at 4°C. Samples were then centrifuged at 14,000g for 25 min at 4° C. The protein concentration in the supernatant was determined by BCA Protein Assay Kit (HyClone-Pierce, Utah, USA). Equivalent amounts (60 μg protein/lane) of protein lysates were separated by 12% SDS-polyacrylamide gel electrophoresis and transferred onto a nitrocellulose membrane (0.22 μm, Invitrogen, NY, USA) in a transfer tank (Bio-Rad, California, USA) using the submerged method. The membrane was blocked for 2 h at room temperature in PBS containing 0.1% Tween-20 (Sigma) and 5% non-fat dried milk (Carnation, city, state, USA), and then incubated with the primary antibody in dilution buffer (1 × TBS, 0.1% Tween-20 with 5% BSA) with gentle agitation overnight at 4°C. Primary antibodies were: anti-BAD (diluted 1:400, rabbit polyclonal), anti-phospho-BAD (Ser136) (diluted 1:200, rabbit polyclonal), anti-Caspase-8 (1C12) (diluted 1:600, mouse monoclonal), anti-caspase-3 (diluted 1:600, rabbit polyclonal, all from Cell Signaling Technology, Beverly, MA, USA), and anti-β-actin (diluted 1:400, mouse monoclonal C-2, Santa Cruz, CA, USA). Then, the membranes were washed in dilution buffer (1 × TBS, 0.1% Tween-20 with 5% BSA) and incubated for 1 hour with horseradish-peroxidase (HRP)-conjugated secondary antibodies, and finally developed by an ECL system (Cell Signaling Technology, Beverly, MA, USA). Secondary antibodies were: HRP-conjugated anti-rabbit IgG (diluted 1:2000) and HRP-conjugated anti-mouse IgG (diluted 1:2000, both from Cell Signaling Technology). The Western blotting was performed following Laemmli’s method [26] and the grayscale values of each band on the blots were measured using BandScan4.3.
Statistical analysis
Data are expressed as the mean ± the standard error of mean (SEM) of separate experiments (n ≥ 3, where n represents the number of independent experiments). All data were tested for significance by one-way analysis of variance (ANOVA) followed by Fisher’s post hoc test using SPSS13.0 software. Results with a P value less than or equal to 0.05 were considered statistically significant.
Results
Cytotoxicity of ardipusilloside III in both U251MG cells and primary astrocytes
The effects of ardipusilloside III on the viability of U251MG cells and primary cultured human astrocytes were assessed by the MTT assay (Fig. 1b). Ardipusilloside III inhibited the growth of U251MG cells in a dose- and time-dependent manner. Viability of U251MG cells treated with 5 μg/ml ardipusilloside III for 6 h decreased to 71.3% (n = 10, P < 0.05). After U251MG cells were treated with 40 μg/ml ardipusilloside III for 72 h, the percent of viable cells significantly decreased to 12.6% (n = 10, P < 0.05). The concentrations at which ardipusilloside III inhibited cell growth by 25% (IC25) and 50% (IC50) at 24 h were 3.7 and 8.2 μg/ml, respectively. However, exposure of the astrocytes to different concentrations of ardipusilloside III (5–40 μg/ml) for 24 h did not result in any statistically significant change in cell viability, with the viability ranging from 81.7% to 92.3% (n = 10, P > 0.05). Therefore, ardipusilloside III reduced the viability of U251MG cells, but was only slightly toxic to the primary cultured human astrocytes.
Cell cycle change in U251MG cells treated with ardipusilloside III
The cellular DNA content was analyzed by flow cytometry to detect changes in the cell cycle distribution. DNA histogram analysis showed that, following incubation with ardipusilloside III at a low concentration (3.7 μg/ml), the percentage of S phase cells reduced from 44.2% to 14.6%; while that of cells in the G2/M phase increased from 8.9% to 26.6% within 48 h, suggesting G2/M phase arrest. At 72 h, the presence of a significant sub-G1 phase (1.4%) fraction suggests induction of apoptosis in the U251MG cells. Ardipusilloside III arrested the cell cycle at the G2/M phase transition prior to the induction of apoptosis in the glioblastoma cells. However, at a higher concentration (8.2 μg/ml), ardipusilloside III quickly induced apoptosis, characterized by the appearance of cells with sub-G1 DNA content, without arresting the cell cycle (Fig. 2a, Table 1).

Effect of ardipusilloside III on cell cycle progression, intracellular structures, phosphatidylserine externalization and DNA fragmentation of human glioblastoma U251MG cells: (a) Flow cytometric analysis of cell cycle phase distribution of U251MG cells after treatment with different concentrations of ardipusilloside III for the indicated time periods; (b) Electron microscopy showed the organization of subcellular organelles of U251MG cells, including the presence of vacuoles containing cytoplasm and nucleolus fragments budding off the plasma membrane (center, magnification ×15,000) and marginal chromatin condensation (right, magnification ×7,500); (c) Representative dot plots showing flow cytometric analysis of glioblastoma U251MG cells treated with different concentrations of ardipusilloside III for the indicated time periods then stained with FITC-conjugated Annexin V and propidium iodide; (d) The characteristic DNA ladder suggesting apoptotic DNA damage in the ardipusilloside III-treated U251MG cells is shown in Lane 3. Cells were treated with 8.2 μg/ml ardipusilloside III for 24 h
Morphological alteration of U251MG cells treated with ardipusilloside III
Hoechst 33342 nuclear staining show that U251MG cells underwent marked morphological changes in a time- and dose-dependent manner after incubation with ardipusilloside III. The U251MG cells shrank, aggregated, and detached from the surface of the culture flask. Apoptotic U251MG cells showed characteristic morphological and biochemical features such as chromatin aggregation, nuclear and cytoplasmic condensation, and the partition of cytoplasm and nucleus into membrane bound-vesicles known as apoptotic bodies (Fig. 3). Electron microscopy revealed the apoptotic changes of intracellular structures in most ardipusilloside III—treated cells. In comparison with the normal intracellular morphology of the control cells, apoptotic U251MG cells lost their microvilli, had increased numbers of lysosomes, and had condensed, fractured, and marginalized chromatin. Also, numerous membrane-enclosed vacuoles containing cytoplasm and parts of the fractured nucleus were found in the treated cells, which would most likey become apoptotic bodies (Fig. 2b).

Morphological alterations of U251MG cells treated with different concentrations of ardipusilloside III for 24 h. Treated cells shrank, aggregated, and detached from the bottom of culture flask as shown in the upper panels (magnification ×600). Nuclear morphology was detected using Hoechst 33342, and visualized by fluorescence microscopy as shown in the lower panels (magnification ×600): (a) Cell condensation and fracturing of marginal chromatin; (b) The lobulated gemmules of the cell nucleus; (c) Apoptotic body
Ardipusilloside III increased phosphatidylserine exposure and induced DNA cleavage in U251MG cells
To quantify early apoptotic effects, the cells were stained with annexin V and propidium iodide (PI) following ardipusilloside III exposure. The right lower quadrant of the density dot plots from the flow cytometric analysis represented early apoptotic cells. Phosphatidylserine externalization occurred after ardipusilloside III treatment in a time- and dose-dependent manner (Table 2). The number of apoptotic cells increased at both concentrations (3.7 and 8.2 μg/ml) with time (6 and 12 h). Despite the fact that the number of early apoptotic cells increased after treatment with the higher concentration, the late apoptotic cells and necrotic cells also increased to some extent, which suggested that treatments should not exceed the IC50, in order to avoid necrosis in vitro (Fig. 2c).
Electrophoresis of cellular DNA revealed a distinctive ladder pattern of DNA cleavage in the apoptotic U251MG cells, which represents the discrete fragments of 180–200 base pairs or multiples thereof that result from non-random cleavage between the nucleosomes. The ladder pattern did not appear in the DNA isolated from control cells (Fig. 2d). Electrophoresis of cellular DNA revealed that ardipusilloside III induced apoptosis specific DNA cleavage in U251MG cells.
Dephosphorylation and cleavage of BAD in ardipusilloside III-induced apoptosis
To determine the involvement of BAD in ardipusilloside III-induced apoptosis, the steady-state levels of phospho-BAD (serine 136) were measured by Western blot analysis (Fig. 4a). The expression of endogenous phospho-BAD was decreased slightly (p-BAD/actin = 89.9% ± 0.4%, n = 3, P < 0.01) after ardipusilloside III treatment for 6 h and decreased even further (78.4% ± 0.1%, n = 3, P < 0.01) after 24 h of treatment, while the ardipusilloside III-free control group showed no difference.

Activation of BAD and caspases involved in ardipusilloside III-induced apoptosis of human glioblastoma U251MG cells: (a) BAD was dephosphorylated at serine136 and cleaved to generate tBADS (15 kDa), as indicated by the arrow. Phospho-BAD expression decreased, while tBADS expression increased, during the indicated time course. The expression of total BAD was slightly decreased after ardipusilloside III treatment. Rapid and substantial caspase-8 and -3 cleavage was detected after treatment with 8.2 μg/ml ardipusilloside III for the indicated time periods; (b) Statistical analysis of the Western blots. Bars, ±SE; *P < 0.05; **P < 0.01
Treatment of U251MG cells with 8.2 μg/ml ardipusilloside III also resulted in Bad cleavage. As early as 6 h after treatment, the tBADS/actin ratio increased significantly (78.8% ± 0.4%, n = 3, P < 0.01) and continued to increase up to 24 h (87.8% ± 0.2%, n = 3, P < 0.01). We detected no cleaved BAD in cells cultured without ardipusilloside III (Fig. 4a). In addition, total BAD expression began to decrease slightly after 12 h of treatment (94.6% ± 0.9%, n = 3, P < 0.01), continuing to decrease up to 24 h (93.6% ± 0.9%, n = 3, P < 0.01).
Western blot analysis well described the dephosphorylation and cleavage of BAD in ardipusilloside III-induced apoptosis, confirmed the intrinsical BAD apoptotic pathway underlying the ardipusilloside III-induced apoptosis.
Caspase-8 and -3 are activated in ardipusilloside III-induced apoptosis
To investigate the contribution of the extrinsic apoptotic pathway in ardipusilloside III-induced apoptosis, we assessed the activation of caspase-8 and caspase-3. Figure 4a shows that, in U251MG cells treated with 8.2 μg/ml ardipusilloside III, both caspase-8 and caspase-3 were rapidly and substantially cleaved. There is some idiopathic activation of caspase-8 in normal glioblastoma cells. However, greatly increased caspase-8 activation was observed after 6 h of treatment (41.7% ± 0.6%, n = 3, P < 0.01) and was maintained (43.6% ± 0.6%, n = 9). The first cleavage products of caspase-3 were found after 6 h of exposure to ardipusilloside III (2.6% ± 1.0%, n = 3, P < 0.05), and increased significantly up to 12 h after exposure (43.5% ± 5.2%, n = 6, P < 0.01). Data suggested the extrinsic apoptotic pathway mediated by caspase-8 and -3 also involved in the ardipusilloside III-induced apoptosis.
Discussion
BAD is regulated primarily by phosphorylation, in response to survival factors, at several sites, including serine 136, which of BAD is vital for 14-3-3 interaction in mammalian cells [27]. BAD phosphorylation protects cancer cells from apoptosis by raising the threshold at which the mitochondria release cytochrome c to induce cell death [28]. Extensive researches have shown that carcinogenesis and cancer progression are highly correlated with BAD phosphorylation [29–31]. Over-expressed phospho-BAD is frequently detected in cancer cells such as rat glioma C6 cells [29], glioblastoma cells [30], and human neuroblastoma SH-SY5Y cells [31]. BAD dephosphorylation and redistribution from the cytosol to the mitochondria are early events in chemotherapy-induced apoptosis, and are regarded as prominent links between the inhibition of survival pathways and the onset of execution phase of apoptosis. Dephosphorylation at serine 136 decreases the accessibility to serine 155 by survival-promoting kinases, disrupting the associations between BAD and prosurvival proteins [32]. Compared to standard treatments, combined usage of anti-cancer agents to prevent BAD phosphorylation could be a promising therapeutic strategy. Recent experiments suggest that many anti-cancer agents, like doxorubicin [16], cannabinoids [30], peroxisome-proliferator-activated receptor-c (PPARc) agonists [33], paclitaxel [34], cyanide [35], and ammonia [36], induce apoptosis by dephosphorylating BAD. We found BAD dephosphorylation in ardipusilloside III-induced U251MG apoptosis. BAD dephosphorylation may trigger the various signaling pathways by which apoptosis is induced in glioblastoma cells.
The final apoptotic pathway examined herein involved the caspase cascades that cleave key cellular components, including cytoskeletal proteins and nuclear proteins, to produce the altered morphology of the treated U251MG cells. Caspases can also activate other catabolic enzymes such as DNases, which cleave DNA into the charcteristic ladder [37] we have observed. Caspases also cleave Bcl-2 family members at specific sites during apoptosis induced by growth factor withdrawal or other apoptotic stimuli [38]. The BAD-caspase positive feedback cascade reaction can amplify the apoptotic signal to some extent. As a response to the cell death signals produced by anticancer agents, BAD is cleaved by caspase-3 to generate two truncated products: tBADL (truncated BAD large, 26 kDa) and tBADS (truncated BAD small, 15 kDa). tBADL inhibits apoptosis in impaired neurons [39]. tBADS is poorly phosphorylated due to alterations in its secondary structure as the result of cleavage, so it interacts inefficiently with 14-3-3 proteins but efficiently with Bcl-xL, thereby altering the Bcl-xL-Bax balance and activating the apoptotic program [39, 40]. Genistein [17], raloxifene [41], and isoliquiritigenin [42] induce apoptosis by cleaving BAD into small fragments in many types of cancer cell; thereby proving that tBADS is a more potent inducer of apoptosis than uncleaved BAD. We have detecetd increased levels of tBADS, which could reinforce the other mechanisms of ardipusilloside III-induced U251MG apoptosis.
Since caspase-3 can be activated directly by caspase-8 in Fas-mediated apoptosis, and glioblastoma cells are Fas-sensitive, tBADS generated from caspase-3-dependent cleavage induces apoptosis more efficiently. Our study also showed that both caspase-8 and -3 were activated immediately after exposure. The activation of caspase-3 was amplified by caspase-8 activation in ardipusilloside III-induced U251MG apoptosis. Therefore, the mechanism of caspase-dependent BAD cleavage may be critical to improving the chemotherapeutic treatment of glioblastoma. However, BAD activation and the caspase cascade occurred at almost the same time, so whether BAD was activated by the caspase cascade or not has not been clearly proven. Herein we have not excluded an independent effect of the caspase cascade on ardipusilloside III-induced apoptosis; however, we consider that both BAD dephosphorylation and the cleavage-mediated intrinsic pathways, as well as the caspase-8 and -3 activation-mediated extrinsic pathways, play an important role in ardipusilloside III-induced apoptosis. In addition, the fact that ardipusilloside III did not significantly affect the viability of astrocytes, together with the neuroprotective effect of tBADL (data not shown), suggests that ardipusilloside III may be safer in a clinical setting than other anti-cancer agents.
Because of the cross regulation necessary for intracellular homeostasis, alteration of one physiological protein may affect more than a single physiological process [15]. Impairment of cell cycle progression may activate the mitochondrial apoptotic pathway [30, 43]. BAD is interesting, both for its apoptosis-regulating capacity and for its effect on cell cycle progression [15]. Our previous study showed that cyclin D2 was overexpressed in glioblastoma cells but hardly expressed in normal brain tissue [44], and was responsible for the progression of glioblastoma cells past the G1/S transition of the cell cycle. It is possible that, following ardipusilloside III treatment, BAD is dephosphorylated and thereby activated, altering the expression of cell cyclins such as cyclin D2, thus leading to cell cycle changes. However, these processes are not yet clear. The time course study of DNA histogram analysis revealed that ardipusilloside III at a low concentration provoked cell cycle arrest at the G2/M phase transition prior to the induction of apoptosis, a higher concentration induced apoptosis without any detectable cell cycle inhibition. These results prove that the apoptotic response of glioblastoma U251MG cells to ardipusilloside III at a low concentration was different from that at a high concentration. Whether U251MG cells undergo cell cycle arrest or apoptosis after exposure to ardipusilloside III at different concentrations is the result of initiation of different pathways. However, whether BAD was involved in the cell cycle arrest needs further study.
In conclusion, adipusilloside III is capable of inhibiting growth and inducing apoptosis of U251MG cells in a dose- and time-dependent manner. The anti-cancer effects of ardipusilloside III may result from multiple mechanisms, such as interfering with cell cycle progression and inducing apoptosis. There are also multiple possible mechanisms by which ardipusilloside III induces apoptosis, through both the intrinsic, BAD dephosphorylation and cleavage pathway, and the extrinsic caspase-8/caspase-3 pathway. We postulate that this new agent may hold a promise as an anti-cancer agent. Based on the observations that ardipusilloside III has significant anti-glioblastoma activity in vitro, further study of its effects and related mechanisms in vivo are needed. This study provides important data to evaluate the possibility of clinical application for ardipusilloside III as an anti-cancer agent.
Conclusion
We identified the novel saponin ardipusilloside III as an anti-cancer and apoptosis-inducing agent using the glioblastoma cell line U251MG cells. Both the BAD-mediated intrinsic apoptotic signaling pathway and the caspase-8-mediated extrinsic apoptotic signaling pathway are involved in ardipusilloside III-induced apoptosis of U251MG cells. These results suggest that ardipusilloside III could be a potential novel agent for use in glioblastoma chemotherapy.
Abbreviations
- IC50 :
-
50% inhibitory concentration
- BAD:
-
Bcl-xL/Bcl-2-associated death promoter homolog
- DMEM:
-
Dulbecco’s modified Eagle’s medium
- MTT:
-
3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide
References
Louis DN, Ohgaki H, Wiestler OD et al. (2007) The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol (Berl) 114(2):97–109
Kita D, Yonekawa Y, Weller M, Ohgaki H (2007) PIK3CA alterations in primary (de novo) and secondary glioblastomas. Acta Neuropathol (Berl) 113(3):295–302
Knobbe CB, Trampe KA, Reifenberger G (2005) Genetic alteration and expression of the phosphoinositol-3-kinase/Akt pathway genes PIK3CA and PIKE in human glioblastomas. Neuropathol Appl Neurobiol 31(5):486–490
Waite DC, Jacobson EW, Ennis FA, Edelman R, White B, Kammer R, Anderson C, Kensil CR (2001) Three double-blind, randomized trials evaluating the safety and tolerance of different formulations of the saponin adjuvant QS-21. Vaccine 19(28–29):3957–3967
Wang QF, Chen JC, Hsieh SJ, Cheng CC, Hsu SL (2002) Regulation of Bcl-2 family molecules and activation of caspase cascade involved in gypenosides-induced apoptosis in human hepatoma cells. Cancer Lett 183(2):169–178
Yun TK (2003) Experimental and epidemiological evidence on non-organ specific cancer preventive effect of Korean ginseng and identification of active compounds. Mutat Res 523–524(54):63–74
Leung KW, Yung KK, Mak NK, Chan YS, Fan TP, Wong RN (2007) Neuroprotective effects of ginsenoside-Rg1 in primary nigral neurons against rotenone toxicity. Neuropharmacology 52(3):827–835
Slovin SF, Ragupathi G, Musselli C, Fernandez C, Diani M, Verbel D, Danishefsky S, Livingston P, Scher HI (2005) Thomsen-Friedenreich (TF) antigen as a target for prostate cancer vaccine: clinical trial results with TF cluster (c)-KLH plus QS21 conjugate vaccine in patients with biochemically relapsed prostate cancer. Cancer Immunol Immunother 54(7):694–702
Green DR, Reed JC (1998) Mitochondria and apoptosis. Science 281(5381):1309–1312
Cheng G, Zhang X, Tang HF, Zhang Y, Zhang XH, Cao WD, Gao DK, Wang XL, Jin BQ (2006) Asterosaponin 1, a cytostatic compound from the starfish Culcita novaeguineae, functions by inducing apoptosis in human glioblastoma U87MG cells. J Neurooncol 79(3):235–241
Chao DT, Korsmeyer SJ (1998) BCL-2 family: regulators of cell death. Annu Rev Immunol 16:395–419
Gleichmann M, Weller M, Schulz JB (2000) Insulin-like growth factor-1-mediated protection from neuronal apoptosis is linked to phosphorylation of the pro-apoptotic protein BAD but not to inhibition of cytochrome c translocation in rat cerebellar neurons. Neurosci Lett 282(1–2):69–72
Thornberry NA, Lazebnik Y (1998) Caspases: enemies within. Science 281(5381):1312–1316
Schendel SL, Xie Z, Montal MO, Matsuyama S, Montal M, Reed JC (1997) Channel formation by antiapoptotic protein Bcl-2. Proc Natl Acad Sci USA 94(10):5113–5118
Maddika S, Ande SR, Panigrahi S, Paranjothy T, Weglarczyk K, Zuse A, Eshraghi M, Manda KD, Wiechec E, Los M (2007) Cell survival, cell death and cell cycle pathways are interconnected: Implications for cancer therapy. Drug Resist Updat 10(1–2):13–29
Grethe S, Coltella N, Di Renzo MF, Porn-Ares MI (2006) p38 MAPK downregulates phosphorylation of BAD in doxorubicin-induced endothelial apoptosis. Biochem Biophys Res Commun 347(3):781–790
Yeh TC, Chiang PC, Li TK, Hsu JL, Lin CJ, Wang SW, Peng CY, Guh JH (2007) Genistein induces apoptosis in human hepatocellular carcinomas via interaction of endoplasmic reticulum stress and mitochondrial insult. Biochem Pharmacol 73(6):782–792
Kim BC, Mamura M, Choi KS, Calabretta B, Kim SJ (2002) Transforming growth factor beta 1 induces apoptosis through cleavage of BAD in a Smad3-dependent mechanism in FaO hepatoma cells. Mol Cell Biol 22(5):1369–1378
Zhang QH, Wang XJ, Miao ZC, Feng R (1993) Studies on the saponin constituents of jiu jielong (Ardisea pusilla). Yao Xue Xue Bao 28(9):673–678
Tao X, Wang P, Yang X, Yao H, Liu J, Cao Y (2005) Inhibitory effect of ardipusilloside-I on Lewis pulmonary carcinoma and hepatocarcinoma SMMC-7721. Zhong Yao Cai 28(7):574–577
Baranes D, Lopez-Garcia JC, Chen M, Bailey CH, Kandel ER (1996) Reconstitution of the hippocampal mossy fiber and associational-commissural pathways in a novel dissociated cell culture system. Proc Natl Acad Sci USA 93(10):4706–4711
Denizot F, Lang R (1986) Rapid colorimetric assay for cell growth and survival. Modifications to the tetrazolium dye procedure giving improved sensitivity and reliability. J Immunol Methods 89(2):271–277
Vindelov L, Christensen IJ (1990) An integrated set of methods for routine flow cytometric DNA analysis. Methods Cell Biol 33:127–137
Gong J, Traganos F, Darzynkiewicz Z (1994) A selective procedure for DNA extraction from apoptotic cells applicable for gel electrophoresis and flow cytometry. Anal Biochem 218(2):314–319
Reynolds JE, Li J, Eastman A (1996) Detection of apoptosis by flow cytometry of cells simultaneously stained for intracellular pH (carboxy SNARF-1) and membrane permeability (Hoechst 33342). Cytometry 25(4):349–357
Laemmli UK (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227(5259):680–685
Masters SC, Yang H, Datta SR, Greenberg ME, Fu H (2001) 14-3-3 inhibits BAD-induced cell death through interaction with serine-136. Mol Pharmacol 60(6):1325–1331
Datta SR, Ranger AM, Lin MZ, Sturgill JF, Ma YC, Cowan CW, Dikkes P, Korsmeyer SJ Greenberg ME (2002) Survival factor-mediated BAD phosphorylation raises the mitochondrial threshold for apoptosis. Dev Cell 3(5):631–643
Ellert MA, Kaminska B, Konarska L (2005) Cannabinoids down-regulate PI3K/Akt and Erk signalling pathways and activate proapoptotic function of BAD protein. Cell Signal 17(1):25–37
Chia CF, Chen SC, Chen CS, Shih CM, Lee HM Wu CH (2005) Thallium acetate induces C6 glioma cell apoptosis. Ann N Y Acad Sci 1042(1):523–530
Li JL, Zhu JH, Jing ZZ, Chen ZC, Xiao ZQ (2003) G-protein-coupled muscarinic acetylcholine receptor activation up-regulates Bcl-2 and phospho-BAD via Ras-ERK-1/2 signaling pathway. Sheng Wu Hua Xue Yu Sheng Wu Wu Li Xue Bao (Shanghai) 35(1):41–48
Datta SR, Katsov A, Hu L, Petros A, Fesik SW, Yaffe MB, Greenberg ME (2000) 14-3-3 proteins and survival kinases cooperate to inactivate BAD by BH3 domain phosphorylation. Mol Cell 6(1):41–51
Zander T, Kraus JA, Grommes C, Schlegel U, Feinstein D, Klockgether T, Landreth G, Koenigsknecht J, Heneka MT (2002) Induction of apoptosis in human and rat glioma by agonists of the nuclear receptor PPARgamma. J Neurochem 81(5):1052–1060
Page C, Lin HJ, Jin Y, Castle VP, Nunez G, Huang M, Lin J (2000) Overexpression of Akt/AKT can modulate chemotherapy-induced apoptosis. Anticancer Res 20(1A):407–416
Shou Y, Li L, Prabhakaran K, Borowitz JL, Isom GE (2004) Calcineurin-mediated BAD translocation regulates cyanide-induced neuronal apoptosis. Biochem J 379(Pt 3):805–813
Yang L, Omori K, Suzukawa J, Inagaki C (2004) Calcineurin-mediated BAD Ser155 dephosphorylation in ammonia-induced apoptosis of cultured rat hippocampal neurons. Neurosci Lett 357(1):73–75
Hengartner MO (2000) The biochemistry of apoptosis. Nature 407(6805):770–776
Seo SY, Chen YB, Ivanovska I, Ranger AM, Hong SJ, Dawson VL, Korsmeyer SJ, Bellows DS, Fannjiang Y, Hardwick JM (2004) BAD is a pro-survival factor prior to activation of its pro-apoptotic function. J Biol Chem 279(40):42240–42249
Condorelli F, Salomoni P, Cotteret S, Cesi V, Srinivasula SM, Alnemri ES, Calabretta B (2001) Caspase cleavage enhances the apoptosis-inducing effects of BAD. Mol Cell Biol 21(9):3025–3036
Cheng AC, Huang TC, Lai CS, Pan MH (2005) Induction of apoptosis by luteolin through cleavage of Bcl-2 family in human leukemia HL-60 cells. Eur J Pharmacol 509(1):1–10
Kim HT, Kim BC, Kim IY, Mamura M, Seong DH, Jang JJ, Kim SJ (2007) Raloxifene, a mixed estrogen agonist/antagonist, induces apoptosis through cleavage of BAD in TSU-PR1 human cancer cells. J Biol Chem 277(36):32510–32515
Jung JI, Lim SS, Choi HJ, Cho HJ, Shin HK, Kim EJ, Chung WY, Park KK, Park JH (2006) Isoliquiritigenin induces apoptosis by depolarizing mitochondrial membranes in prostate cancer cells. J Nutr Biochem 17(10):689–696
Konishi Y, Lehtinen M, Donovan N, Bonni A (2002) Cdc2 phosphorylation of BAD links the cell cycle to the cell death machinery. Mol Cell 9(5):1005–1016
Zhang X, Zhao M, Huang AY, Fei Z, Zhang W, Wang XL (2005) The effect of cyclin D expression on cell proliferation in human gliomas. J Clin Neurosci 12(2):166–168
Acknowledgments
The authors would like to thank Juan Li for her excellent technical assistance and Rui-Feng Cao for his helpful discussions. This work was funded by Chinese National Natural Science Foundation, Grant number: 20502035.
Author information
Authors and Affiliations
Corresponding author
Additional information
Hong Lin, Xiang Zhang, Guang Cheng, and Hai-Feng Tang contributed equally to the work.
Rights and permissions
About this article
Cite this article
Lin, H., Zhang, X., Cheng, G. et al. Apoptosis induced by ardipusilloside III through BAD dephosphorylation and cleavage in human glioblastoma U251MG cells. Apoptosis 13, 247–257 (2008). https://doi.org/10.1007/s10495-007-0170-9
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10495-007-0170-9