
Abstract
Oribatid mites are important decomposers of dead organic matter in soils across the world. Their origin dates back at least 380 Mya. Multiple severe climatic changes during Late Pliocene and Pleistocene shaped the migration patterns of these organisms and should be reflected in the genetic variability of their current populations. In this study, we examined the genetic diversity and phylogeographic structure as well as the evolutionary history of populations of two ecologically different oribatid mite species. Pantelozetes cavaticus is a troglophile oribatid mite known mainly from Central European caves, whereas Pantelozetes paolii is a common surface eurytopic species with Holarctic distribution. We used two molecular markers—mitochondrial cytochrome c oxidase subunit I (COI) and the nuclear D3 region of the 28S rDNA gene—to reveal phylogenetic relationships between contemporary populations. Whereas the D3 region showed minimal or no variability within populations, COI appeared to be a relevant marker for population studies. Phylogeographic analysis based on COI detected two lineages of P. cavaticus (‘Czech’ and ‘Slovak’), which separated during the Late Pliocene (2.9 Mya) and revealed the existence of one new species. In contrast, three identified genetic lineages of P. paolii (radiation time 2.9 and 1.2 Mya, respectively) uncovered in this study were found to coexist in the distant sampling localities, suggesting a connection between populations even over long distances.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Oribatid mites (Acari: Oribatida) are mostly a soil-dwelling and very diverse group of microarthropods with more than 11,000 species described so far (Subías 2020). Their representatives can be found in almost every terrestrial habitat from tropics to poles, being the most abundant group in the first few centimeters of soil, with a density up to hundreds of thousands of individuals per m2 in temperate forests with a thick humus layer (Maraun and Scheu 2000). They play an important functional role in soils as decomposers of dead organic matter and stimulators of fungal and bacterial growth (Luxton 1972).
Oribatid mites are among the oldest terrestrial animals, with earliest fossil record from Devonian sediments dated at 380 Mya (Norton et al. 1988) and molecular analyses dating their origin to at least 580 Mya (Schaefer et al. 2010). In Europe, climatic oscillations and consequential biome changes over the past 3 million years caused considerable shifts in distributional patterns of surface as well as soil-living animals (Hewitt 2004). During the cold periods, the warm-adapted fauna and flora either went extinct or were forced to look for more favourable habitats, i.e., refugia. However, active dispersal skills of the soil mesofauna, such as Oribatida or Collembola, are quite limited (Lehmitz et al. 2012) and more recent climatic and habitat changes during Pliocene and Pleistocene defined the migration patterns of the individual species, which should be still reflected in the genetic composition of their current populations. Such effects on the genetic diversity have been studied only scarcely in soil-dwelling arthropods (Beheregaray 2008; Rosenberger 2010), although present-day distributional patterns of intraspecific genetic diversity and estimations of its degree have been shown to provide important insights into the phylogeography and evolutionary history of species (Beebee and Row 2008).
Yosii (1956) pointed out that caves with constant microclimatic conditions in the northern temperate zone can serve as refugia, especially for small soil-dwelling species. Subterranean ecosystems are generally considered as habitats where species from different external ecological conditions can successfully cohabit during hostile surface conditions (Kováč et al. 2016).
The troglophile Pantelozetes cavaticus (Kunst) is a sexually reproducing oribatid mite that has been reported mainly from Central European caves, in eastern Czech Republic (Starý 2008), in the Western Carpathians and the Slovak Karst in Slovakia (Ľuptáčik and Miko 2003), in eastern Austria (Bruckner 1995), in southern Poland (Żbikowska-Zdun et al. 2009) and in some caves in Germany, Belgium and Hungary (Ľuptáčik 2004). The species does not possess any obvious troglobiomorphic adaptations (e.g., depigmentation, elongated antennae and legs), but it shows a strong affiliation to the cave environment. Only two findings have been reported from surface environments (Starý 2008; Żbikowska-Zdun et al. 2009). It is considered coprophilous (guanophilous) with frequent occurrence on rotten wood (Ľuptáčik and Miko 2003). The distribution pattern along with absent adaptations to cave life of P. cavaticus indicates that it could be a glacial relict, most likely a descendant of an old Pleistocene fauna, or even older Tertiary fauna.
Żbikowska-Zdun et al. (2009) examined populations of P. cavaticus from two isolated caves in Poland and found no significant morphological differences between them. Here we take an approach of investigating possible differences between populations at the molecular level. Deep genetic divergences in soil-dwelling arthropods at the population level have been previously reported and well documented in oribatid mites (Heethoff et al. 2007; Rosenberger 2010; Saltzwedel et al. 2014; Schäffer et al. 2019).
To evaluate whether the association of P. cavaticus with subterranean habitats caused a geographical isolation and consequently a reproductive isolation, we compared its genetic variability with that of populations of a closely related surface species Pantelozetes paolii (Oudemans). In contrast to P. cavaticus, P. paolii is a widespread and abundant eurytopic pan-phytophagous species with sexual reproduction and Holarctic distribution. Pantelozetes paolii is a type species of the genus Pantelozetes, which now comprises 22 species and one subspecies based on morphological characters (Subías 2020); phylogenetic relationships among these species are unknown.
In the present study, we used the combination of two DNA markers—the mitochondrial cytochrome c oxidase subunit I (COI, the barcoding region) and the nuclear D3 region of the 28S rDNA gene—as both markers were previously successfully used in other studies aimed at phylogeny and phylogeography of oribatid mites (COI: Rosenberger 2010; Schäffer et al. 2010, 2019; Pfingstl et al. 2019; D3: Lehmitz and Decker 2017; Pachl et al. 2017). Furthermore, Kreipe et al. (2015) suggested that the combination of these two genes should provide a reliable insight into the phylogeny and species radiation within oribatid mites.
Our objectives were (1) to investigate the genetic variability within and between the populations of the troglobiotic P. cavaticus and to determine whether the studied caves were reproductively isolated; (2) to compare the genetic variability of two congeneric oribatid mite species with a different ecology—P. cavaticus and P. paolii; and (3) to trace the evolutionary history of potential lineages of both species. Assuming that the active dispersal abilities of oribatid mites are limited, we expected similar patterns of genetic variability, with genetic isolation more pronounced in the troglobiotic species even over shorter geographical distances.
Materials and methods
Specimen sampling, DNA extraction and sequencing
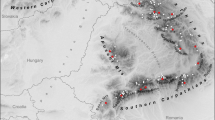
Organic litter, decaying wood or bat guano samples for the extraction of P. cavaticus were taken from sediments in 10 caves between 2015 and 2017 (three caves in Czech Republic, seven in Slovakia; see Fig. 1), where the presence of this species was demonstrated in previous research (Ľuptáčik 2004; Starý 2008). Nová Amatérská Cave and Sloupsko-Šošůvské Caves are in the Moravian Karst, which is the largest karst area in Czech Republic based in the Middle Devonian limestone. These two cave systems are connected via a subterranean stream. A third cave sampled in Czech Republic, the Javoříčské Caves, is situated in a small outcrop of Devonian limestone in the Špraněk Hill. Five of the caves sampled in Slovakia are part of the Slovak Karst (the largest karst area in Central Europe composed of several layers of Mesozoic limestone and dolomite) and are relatively close to each other—500 m to 40 km. Belianska Cave is the largest cave in the Slovak part of the Tatra Mountains established in Mesozoic limestone. The last cave sampled in Slovakia, Andrejová Cave II, located in the Čierna Hora Mountains (part of the Slovak Ore Mountains), is a small joint cave in a limestone rock cliff.

Map of Czech Republic, Slovakia and Poland with the sampling localities; dots for Pantelozetes cavaticus and stars for P. paolii (for detailed information about each sampling locality see Table 1, Online Resource 1)
For the common soil living P. paolii, soil samples to the depth of 5 cm were collected at 12 already established study sites of the Institute of Soil Biology (Biology Centre CAS, České Budějovice) between 2016 and 2017 (Czech Republic, Slovakia, and Poland; Fig. 1).
The name of the sampling locality and its abbreviation, number of collected specimens as well as the number of sequences obtained for each locality and GenBank reference numbers are provided for all records in Table 1. More detailed information about sampling localities (GPS, altitude, material collected, etc.) are given in Online Resource 1.
Soil arthropods were extracted in a high-gradient extractor by Marshall (1972) for 5 days with increasing temperatures. Pantelozetes cavaticus and P. paolii specimens were identified to species level under a light microscope after Weigmann (2006) and stored in 96% ethanol at −20 °C until preparation.
Genomic DNA was extracted from single whole individuals (20 specimens of P. cavaticus and 10 of P. paolii from every sampling locality if such a number was available—see Table 1 for details) using the Exgene Tissue SV mini kit following the manufacturer’ protocol for insects with final elution in 50 µl instead of 200 µl (GeneAll® Biotechnology). A fragment of cytochrome c oxidase subunit I (COI) was amplified using the primers OriLCO (5′-TCAACAAATCATAAAGAYATYGG-3′; slightly modified primer COIarchI used in Heethoff et al. 2007), and standard HCO2198 (5′-TAAACTGGGTGACCAAAAAATCA-3′; Folmer et al. 1994). For the amplification of the D3 fragment of nuclear 28S rDNA gene, the forward primer D3A and the reverse primer D3B were used as described in Kreipe et al. (2015).
The polymerase chain reaction (PCR) contained 0.75 µl of each primer (0.5 pmol/µl), 1 µl of dNTPs (2.5 mM of each dNTP), 1.25 µl of 10× reaction buffer, 0.1 µl of TaKaRa Ex Taq® polymerase, 7.75 µl of PCR water and 2 µl (for COI) and 1 µl (for D3), respectively, of template DNA. The PCR conditions consisted of initial 94 °C for 1 min; 40 cycles at 94 °C for 15 s, 47 °C for 40 s and 72 °C for 50 s; and final extension at 72 °C for 2 min. PCR products were visualized on 2% agarose gel and samples with positive products were enzymatically purified with a mixture of 0.5 µl Exonuclease I, 1 µl FastAP (ThermoFisher Scientific, USA) and 0.5 µl PCR water per sample (incubated at 37 °C for 30 min followed by 15 min at 80 °C) before direct sequencing. The purified PCR products were sequenced by GATC Biotech (Germany).
A surface-dwelling oribatid mite Nothrus silvestris (Nicolet) (Crotonioidea) was used as an outgroup taxon. Its COI and D3 sequences were taken over from Rosenberger (2010) and Lehmitz and Decker (2017), respectively (GenBank acc. nr. JF263835 and KY681356, respectively).
All sequences generated for this study were checked to be consistent with oribatid mite DNA via Blast searches (Altschul et al. 1997); no contaminations were discovered. All sequences generated for this study are publicly available from GenBank.
Data analysis
Sequences were manually edited: ambiguous positions were corrected by hand and unreadable short stretches were trimmed using the chromatograms (ca. 30 bp at the 5′ and 3′ ends) with BioEdit v.7 (Hall 1999). Both COI and D3 sequences together with the outgroup taxon N. silvestris were aligned with the MEGA X software (Kumar et al. 2018) by Muscle algorithm with default parameters. The final alignment of the COI fragment contained 203 sequences with the length of 630 bp for P. cavaticus and 63 sequences with the length of 626 bp for P. paolii, respectively. The final alignment of the D3 gene fragment contained 100 sequences (462 bp) for P. cavaticus and 60 sequences (456 bp) for P. paolii. Mitochondrial gene sequences were translated into amino acid sequences using the Invertebrate Mitochondrial Gene Code implemented in MEGA and as there were no stop codons and the alignments were gap-free, all of them were considered as true mitochondrial and not nuclear copies and were reverse-translated into nucleotide sequences for further analyses. All alignments are available from the authors upon request.
The number of synonymous and non-synonymous mutations in the COI alignment were calculated in MEGA, nucleotide (π) and haplotype diversity (Hd) were calculated in DnaSP v.5.10 (Librado and Rozas 2009). The number of protein haplotypes was determined using the online tool DNAcollapser in FaBox v.1.5 (Villesen 2007). The independent analysis of molecular variance (AMOVA) was performed only for P. cavaticus in ARLEQUIN v.3.5 (Excoffier et al. 2005), to investigate within- and between-populations structure based on uncorrected p-distances selecting Czech Republic and Slovakia as groups. Isolation by distance was tested by Mantel Test (10,000 permutations) implemented in ARLEQUIN.
To measure the effect of demographic changes on the DNA sequences of the populations, the neutrality tests Tajima’s D (Tajima 1989) and Fu’s FS (Fu 1997) were performed in ARLEQUIN. Haplotype networks were constructed in PopART v.1.7 (Leigh and Bryant 2015) using Median Joining Network, separately for P. cavaticus and P. paolii, to determine and visualize the relationships and history between haplotypes.
The best-fitting model for phylogenetic analyses of COI alignments of both species was selected using the Bayesian Information Criterion (BIC) in the ModelFinder (Kalyaanamoorthy et al. 2017) implemented in IQ-TREE v.1.6.12 (Nguyen et al. 2015). The model of sequence evolution was HKY + F + G4 for P. cavaticus and HKY + F + I for P. paolii, respectively. Phylogenetic trees of both species were calculated separately using Maximum Likelihood (ML) and Bayesian Inference (BI) methods, implementing the models selected by ModelFinder. ML analyses were conducted in IQ-TREE with default parameter settings and 10,000 ultrafast bootstrap replicates (Hoang et al. 2018). BI was conducted in MrBayes v.3.2.7 (Ronquist et al. 2012) and BEAST v.2.6.0 (Bouckaert et al. 2019). The Bayesian Markov Chain Monte Carlo simulations in MrBayes were performed in two independent runs with four chains for each species separately, each for 10 million generations sampled every 1000th generation, a burn-in of 2500 was applied. To assess run convergence, the Tracer v.1.7.1 (Rambaut et al. 2018) was used.
Molecular divergence times of major lineages were estimated separately for P. cavaticus and P. paolii. Prior to this analysis, substitution saturation of the COI sequences was measured in DAMBE 6 software (Xia 2017) with the test by Xia et al. (2003)—the sequences experienced little substitution saturation. The relaxed clock log-normal analysis was performed in two runs for both species with BEAUti, BEAST and TreeAnnotator, all v.2.6.0 (Bouckaert et al. 2019). We set a fixed substitution rate of 0.0115, which corresponds to a standard arthropod mutation rate of COI, 2.3% sequence divergence per million years (Avise 1994; Brower 1994). The site model was HKY + F + G4 for P. cavaticus and HKY + F + I for P. paolii, respectively, and ‘Coalescent Constant Population’ was used as tree prior (as recommended for population-level studies by the authors of BEAST). The population size was set as log normal; all the other priors were estimated by the software. The convergence of the MCMC chain after 10,000,000 generations with every 1000th generation sampled and a burn-in of 1000 was confirmed using the Tracer.
Results
DNA extraction and subsequent PCR amplification of the COI gene was successful in 203 out of 212 P. cavaticus specimens and in 63 out of 157 P. paolii specimens analysed (Table 1); various sets of primers and PCR conditions were tested on the problematic individuals, albeit without success.
PCR amplification of the D3 region worked for all processed samples; however, it showed only a very low molecular variation between the two studied species (uncorrelated p-distances were 0.6%), and none within them. Only four positions of the 462 bp fragment varied in the 100 analysed individuals of P. cavaticus and no position of the 456 bp fragment varied in the 60 analysed individuals of P. paolii. Accordingly, the phylogenetic trees had no structure and sampling localities were mixed (ML phylogenetic tree of studied species together with an outgroup based on the D3 is shown in Online Resource 2a). Therefore, D3 sequences were not used for further analyses.
Phylogenetic analysis of COI sequences split the analysed species into three lineages and suggested the existence of a new species independent of P. cavaticus and P. paolii—hereafter it is referred to as Pantelozetes sp. The new species showed a closer relationship to P. cavaticus, values of uncorrected p-distances in the COI gene were 20.4% for P. cavaticus vs. Pantelozetes sp. and 22.7% between P. paolii and Pantelozetes sp. (Table 2a). D3 sequences failed to distinguish Pantelozetes sp. from P. cavaticus. Pantelozetes sp. was found only at the sampling locality Andrejová Cave II. Subsequent re-inspection under the light microscope revealed distinctive morphological features of the new species and its detailed description is currently in progress and will be published separately. Intraspecific p-distances were 0.2% and five haplotypes were detected in the 23 analysed Pantelozetes sp. COI sequences, which were not used for the reconstruction of phylogenetic trees and haplotype network of the studied species. ML phylogenetic tree of all studied species (P. cavaticus, P. paolii, Pantelozetes sp.) with an outgroup N. silvestris is shown in the Online Resource 2b.
Pantelozetes cavaticus
In each of the phylogenetic trees P. cavaticus was monophyletic and separated with high support from the outgroup taxon N. silvestris (as well as from P. paolii and Pantelozetes sp.; Online Resource 2b). Phylogenetic reconstructions based on BI and ML methods of the COI nucleotide alignment revealed very similar topologies with slightly different resolution. Only the BI tree (created in BEAST) is shown in Fig. 2a (see Online Resource 3 for the other trees). COI haplotypes generally clustered according to sampling locations and separated with high support into two main lineages—‘Czech’ (1) and ‘Slovak’ (2) (Fig. 2a). Applying a standard invertebrate mitochondrial substitution rate of 2.3% per million years, ‘Czech’ and ‘Slovak’ lineages diverged in the Late Pliocene, 2.9 ± 1.6 Mya (Fig. 2a).

Bayesian inference trees based on the COI gene showing the relatedness among individuals of (a) Pantelozetes cavaticus (calculated in BEAST) and (b) P. paolii (calculated in MrBayes). Different branch colors indicate genetic lineages identified within the species (see color version online). Numbers at the nodes represent posterior probabilities, bold numbers are median estimated divergence times ± 95% HPD (highest posterior density) calculated in BEAST
The COI nucleotide haplotype network also showed a strong cave-related structure once more with an obvious separation of Czech and Slovak caves (Fig. 3a). Individuals from nearby located sampling localities (caves) shared identical or closely related haplotypes. Haplotypes of the individuals from Belianska Cave (SK) and Javoříčské Caves (CZ) were clearly separated and not shared with any other individual from another caves. In total, 37 nucleotide haplotypes were sampled within the 180 individuals sequenced. The estimated haplotype diversity was relatively high within the populations and lineages (average values 0.502 and 0.802, respectively), whereas nucleotide diversity was considerably lower in both cases (on average 0.004 in populations and 0.009 in lineages) (Table 3). As for the amino acid haplotypes, 16 were identified, two (most abundant) shared among the individuals from Czech and Slovak caves, while the remaining 14 were detected only in Slovak caves (ML phylogenetic tree based on amino acid sequencies is shown in the Online resource 4a).

Median Joining Haplotype Network of COI sequences of (a) Pantelozetes cavaticus and (b) P. paolii (see color version online). The size of the circles is proportional to the number of sequences per haplotype, bars on the lines represent number of mutation steps separating haplotypes
Genetic distances between the populations from discrete caves were moderately higher and ranged between 0.4 and 4.8% for the COI gene (Table 2b). Within-population genetic distances were generally low (Table 2b). Accordingly, as indicated by AMOVA, genetic variance was highest between groups (Czech and Slovak caves: 70.5%), markedly lower among populations within groups (21.1%) and lowest within populations (8.5%) (Table 4). A significant correlation between genetic and geographical distances of populations, isolation by distance, was revealed using Mantel test (R2 = 0.877, p = 0.004). The neutrality analyses did not show any significant differences from zero within the populations (p > 0.05 for both D and FS), except for two caves—Majkova Cave and Sniežná diera Cave—indicating a population expansion after a bottleneck event (Table 3).
Pantelozetes paolii
The phylogenetic trees based on BI and ML methods of the COI nucleotide alignment showed similar topologies and only the BI tree (MrBayes) is shown in Fig. 2b (ML tree is shown in Online Resource 5). Pantelozetes paolii was monophyletic and separated with high support from the outgroup taxon N. silvestris (as well as from P. cavaticus and Pantelozetes sp., Online Resource 2b) and the COI haplotypes clustered into three main lineages independent of the sampling site (Fig. 2b; for detailed information about the number and geographic origin of the individuals from each lineage see Online resource 6). Applying a mitochondrial substitution rate of 2.3% per million years, the lineage 1 diverged from the rest 2.9 ± 1.1 Mya during the Late Pliocene, and lineages 2 and 3 split during the Pleistocene 1.2 ± 0.6 Mya (Fig. 2b).
The COI haplotype network showed a clear structure of three dominant haplotypes, i.e., three lineages shared among individuals from often very distant sampling localities (Fig. 3b). The network showed individuals from multiple sampling localities in all lineages, i.e., in the sites Ružín Water Dam (RP_SK), NP Krkonoše (KR_CZ) and Jeseníky PLA (JE_CZ) individuals with all three haplotypes coexisted, and in the rest of the sampling sites, a mix of two of the three main haplotypes was found. In total, 12 haplotypes were found in the 63 individuals sequenced. The most abundant haplotypes 1 and 2 were associated with some other haplotypes (represented only by one individual) by a mutational step. Contrary to P. cavaticus, the estimated haplotype diversity was relatively high within the populations (an average value 0.545) but much lower within the lineage (an average value 0.198), whereas nucleotide diversity was low in both cases (and markedly lower within the lineages) (Table 3). We identified six amino acid haplotypes, two abundant: first shared only between the individuals from lineage 1, second shared between individuals from lineages 2 and 3; the rest of the amino acid haplotypes were detected only in one individual on different sampling localities (ML phylogenetic tree is shown in Online Resource 3b).
Genetic distances detected between populations from discrete sampling localities were high and ranged between 1.9 and 5.7% for the COI gene. Intrapopulation genetic distances in the COI gene were high as well (3.3–5%), except for two populations where the value was zero (possibly the result of the small number of successfully sequenced individuals). After redefining the groups of individuals according to their assignment to the genetic lineage, the genetic distances among the lineages ranged between 2.6 and 7% and between 0 and 0.1% within the lineages, respectively (Table 2c).
Isolation by distance was rejected, Mantel test being not significant (R2 = 0.034, p = 0.14). The neutrality analyses did not show any significant differences from zero within the populations (p > 0.05 for both D and FS) leading to the assumption that there is no evidence of size changes in the populations. However, after this analysis was carried out on every identified lineage (except lineage 3 with only one haplotype), values significantly different from zero were observed (p > 0.05 for both D and FS) indicating that the lineages may have undergone a process of population expansion after a bottleneck event (Table 3).
Discussion
The results of our study provide the first insight into the genetic diversity and population structure of two oribatid mite species from the genus Pantelozetes. In agreement with our hypothesis, the cave-associated species, P. cavaticus, is represented by several phylogeographically subdivided populations that are reproductively well isolated between discrete karst areas, falling into two main genetic lineages (‘Czech’ and ‘Slovak’). Conversely, the other investigated species, the common soil-living P. paolii, lacks any geography-related population structure, i.e., the three identified main genetic lineages coexist on individual sampling locations, suggesting good dispersal abilities of this species.
The intraspecific variation of the COI gene was quite high between the identified lineages of both species (ranging from 2.6 to 7%). The standard COI barcoding gene shows in general more variability between the populations of flightless or less mobile wide-spread taxa (Papadopoulou et al. 2009), which was already documented also for the populations of soil-living microarthropods (Heethoff et al. 2007; Schäffer et al. 2010; Rosenberger et al. 2013; Kreipe et al. 2015; Lehmitz and Decker 2017) and which corresponds with our results. The phylogenetic analysis of P. cavaticus COI sequences further revealed existence of new species Pantelozetes sp., which was clearly separate from P. cavaticus (20.4% interspecific distance). A morphological re-inspection of the samples from a small, isolated locality in Čierna Hora Mts. (Andrejová Cave II), where the genetic analysis indicated the presence of the new species, revealed individuals with minor yet distinctive morphological features.
The D3 region was considered as a possible species marker in several studies (Maraun et al. 2003; Laumann et al. 2007; Lehmitz and Decker 2017), although some have reported it to fail to separate closely related species (Lehmitz and Decker 2017; Schäffer et al. 2019). In our study we found only little variation in D3 between P. cavaticus and P. paolii (0.6%); furthermore, the locus also failed to distinguish the new species, with its sequences being identical to those of P. cavaticus. It seems that the D3 fragment is too short and conserved to be reliably used as the sole species marker for oribatid mites.
Pantelozetes cavaticus
The phylogeographic analysis of P. cavaticus sampled in the mid-point of its distribution revealed deep genetic differences between the individual populations. Considering the limited active migration possibilities of the species and, more importantly, the discontinuous nature of the cave environment to which the species is bound, the geographic isolation was expected. Two main lineages of P. cavaticus were identified, ‘Czech’ and ‘Slovak’. Both lineages had specific, non-synonymous substitutions in the COI gene and were never found to coexist in a single cave, indicating selection followed by a subsequent spread of the most competitive genotype. This is consistent with the results of several other studies that revealed high intraspecific genetic diversity in COI of widespread oribatid mite species (Rosenberger 2010; Schäffer et al. 2010, 2019; von Saltzwedel et al. 2014; Pfingstl et al. 2019).
Mutation rate of the mitochondrial COI gene is relatively high, which could lead to the formation of a significant inter-population diversity over a short evolutionary timescale (Hebert et al. 2003). Therefore, the presence of individuals from different caves that shared the same haplotypes (i.e., AM_CZ + SSJ_CZ; LD_SK + MJ_SK + CD_SK; SD_SK + MR_SK) points to a continual gene flow between these populations or to a recent colonization of one cave from the other. This could be easily explained by a short geographical distance between these caves and their potential connection via underground streams or crevices (e.g., Nová Amatérská Cave and Sloupsko-Šošůvské Caves are connected by a subterranean stream, and all the caves from Slovak Karst are close to each other).
In contrast, no shared haplotypes and presumably no gene flow between the populations from distant and not inter-connected karst areas indicate effective reproductive isolation of these populations from each other. A very similar trend of COI genetic variability increasing with geographical distance was detected by Parimuchová et al. (2017) in the populations of the troglophile collembolan Protaphorura janosik in Slovak caves.
Molecular divergence estimates based on P. cavaticus COI sequences indicated that the separation of ‘Czech’ and ‘Slovak’ lineages substantially predated Quaternary glaciations (Pleistocene) and happened during the Late Pliocene (2.9 Mya). This radiation event coincides with the climatic and biotic changes occurring in Europe during this epoch, i.e., global cooling, less precipitation, and the spread of grasslands (Retallack 2001). This might have forced the common ancestor of extant P. cavaticus lineages to escape from the changing surface conditions to cave ecosystems with more stable conditions. Despite this relatively ancient radiation, the lineages have remained morphologically consistent and no clear morphological differences were evident between individuals during the identification prior to the DNA extraction. The stable conditions of the subterranean environment may have contributed to the morphological consistency of the emerging genetic lineages. Homogenous habitat conditions were suggested to enforce stabilizing selection, which can maintain a constant phenotype across the range of the group, resulting in conserved morphologies on a diverse genetic background (Colborn et al. 2001; Pfingstl et al. 2019).
Nevertheless, long-term geographical and genetic isolation of the lineages could be expected to provoke, through the growing genetic distances, an evolution of new species differing even in their morphology. Indeed, our discovery of a new species in Andrejová Cave II, seems to support this theory. Based on the molecular divergence estimates, Pantelozetes sp. separated from the ancestor of P. cavaticus 10.1 (± 1.8) Mya, during the Late Miocene.
Pantelozetes paolii
Pantelozetes paolii is a eurytopic and abundant species that can be found in a variety of surface habitats (with Holarctic distribution), indicating that this species can cope with a wide range of environmental conditions. Unfortunately, the number of specimens obtained during sample collection was limited and PCR amplification of the COI gene did not work very well in this species, therefore the number of successfully sequenced individuals from some sampling localities was low (i.e., only three individuals from DV_CZ and VT_SK).
We assumed that given its small size and limited active locomotion powers, the populations from distant areas would create a distinct phylogeographic pattern as this was already described in many other studies investigating the genetic structure of populations of small widespread soil arthropods (Schäffer et al. 2010; Rosenberger et al. 2013; Saltzwedel et al. 2016, 2017). Waters et al. (2013) suggested that a founder effect may play a major role in the colonization of new habitats, which results in a low genetic variance within the populations but a high one between them.
Contrary to this, in 63 sequenced individuals we found three main genetic lineages with deep genetic differences (genetic distances between the lineages ranged from 2.6 to 7%) cohabiting at distinct sampling localities. High haplotype diversity indicated ancient separation and independent evolution of these lineages. It was common that individuals from at least two lineages (and in a few cases even all three—in Ružín Water Dam, Jeseníky PLA and NP Krkonoše) coexisted at one locality (one haplotype was shared among the individuals from distinct localities). This points to an effective long-distance dispersal ability of this species.
Poor active dispersal, even on distances of only couple of centimeters, is characteristic for oribatid mites (Lehmitz et al. 2012). However, given that many species have huge geographical distribution ranges, efficient dispersal pathways must exist. This topic has not yet been completely resolved. Passive dispersal by wind, though documented, is species-specific and not very common over a longer distance in oribatid mites given their small size and susceptibility to dehydration (Lehmitz et al. 2011; Schuppenhauer et al. 2019). Transport by running water and on larger animals, especially on birds, is more common and well documented (Lebedeva and Krivolutsky 2003; Krivolutsky and Lebedeva 2004; Schuppenhauer et al. 2019). Pantelozetes paolii has been sporadically found in the feathers of waterfowl (Krivolutsky and Lebedeva 2004), which is probably the most likely dispersal manner of this species.
High haplotype diversity (Hd, on average 0.545 and 0.198 for populations and lineages, respectively) and relatively low nucleotide diversity (π, on average 0.035 and 0.001 for populations and lineages, respectively) of P. paolii obtained in this study is consistent with what has been described for some other species of oribatid mites. For example, Schäffer et al. (2010), in their analysis of the COI region of two Scutovertex species (sampled also in Central Europe), observed values of Hd between 0.818 and 0.989 and values of π between 0.018 and 0.051.
These differences in Hd and π can indicate recent population growth (Korstian et al. 2015), which is consistent with the results of the neutrality tests performed within the detected lineages of P. paolii in our study. It seems that the species experienced an event that caused a drastic reduction in its abundance (bottleneck effect) followed by a rapid population growth. First of these events might have happened during the Late Pliocene (2.9 Mya per molecular divergence time estimates) when the climatic and habitat conditions were rapidly changing—similarly to the situation in P. cavaticus as described above. The second event of radiation (1.2 Mya) was probably caused by strong climatic oscillations during the Pleistocene Epoch (Hewitt 2004). Moreover, bottleneck events followed by demographic expansions have been consistently shown to leave a genetic mark in the current populations in the pattern of some haplotypes being broadly shared and others, less frequent, differing by only a few mutations (Bas 1995).
Initial morphological examination of specimen did not reveal substantial differences, however an in-depth morphometric study of individuals from different genetic lineages may reveal relevant morphological traits. Additional coupled genetic and morphological data from representative sampling sites across the P. paolli distribution range are necessary to clarify the relationships among its populations.
Conclusions
This study provides the first insight into the genetic diversity, population structure and evolutionary history of two ecologically different species from the genus Pantelozetes. We show that populations of the cave-dwelling P. cavaticus are effectively confined to the respective karst areas, whereas the populations of the surface-dwelling P. paolii from comparably distant localities are intermixed, demonstrated as three main lineages that coexist on multiple sampling sites, suggesting effective long-distance dispersal of the latter species. The estimated divergence of the genetic lineages of P. cavaticus and P. paolii of 2.9 Mya coincides with the onset of major climatic changes during Late Pliocene, when the European climate began to cool rapidly. In addition, we confirmed that COI is a good marker for studies of population structure, but found the D3 fragment too conserved to distinguish populations or even closely-related species. Finally, we report a new candidate species, Pantelozetes sp., from one of the sampled caves in Slovakia (Andrejová Cave II); its description will be published separately.
Data availability
All the sequences obtained for this study are publicly available from the GenBank (accession numbers are listed in Table 1). The datasets (alignments) generated and analyzed during the current study are available from the corresponding author on reasonable request.
References
Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25(17):3389–3402
Avise JC (1994) Molecular markers, natural history and evolution. Chapman & Hall, New York
Bas C (1995) Ecological structures: expansion and replacement. Sci Mar 59:373–380
Beebee T, Row G (2008) An introduction to molecular ecology, 3rd edn. Oxford University Press, Oxford
Beheregaray LB (2008) Twenty years of phylogeography: the state of the field and the challenges of the southern hemisphere. Mol Ecol 17(17):3754–3774
Bouckaert R, Vaughan TG, Barido-Sottani J, Duchêne S, Fourment M, Gavryushkina A et al (2019) BEAST 2.5: an advanced software platform for Bayesian evolutionary analysis. PLoS Comput Biol 15(4):e1006650
Brower AVZ (1994) Rapid morphological radiation and convergence among races of the butterfly Heliconius erato inferred from patterns of mitochondrial DNA evolution. PNAS USA 91:6491–6495
Bruckner A (1995) Cave-dwelling oribatid mites (Acarina, Cryptostigmata) from East Austria. Verh Zool-Bot Ges Österr 132:81–107
Colborn J, Crabtree RE, Shaklee JB, Pfeiler E, Bowen BW (2001) The evolutionary enigma of bonefishes (Albulaspp.): cryptic species and ancient separations in a globally distributed shorefish. Evolution 55(4):807–820
Excoffier LG, Laval G, Schneider S (2005) Arlequin ver. 3.0: an integrated software package for population genetics data analysis. Evol Bioinform Online 1:47–50
Folmer O, Black M, Hoeh W, Lutz R, Vrijenhoek R (1994) DNA primers for amplification of mitochondrial cytochrome C oxidase subunit I from diverse metazoan invertebrates. Mol Mar Biol Biotechnol 3(5):294–299
Fu YX (1997) Statistical tests of neutrality of mutations against population growth, hitchhiking and background selection. Genetics 147(2):915–925
Hall TA (1999) BioEdit: A User-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acid Symp Ser 41:95–98
Hebert PDN, Cywinska A, Ball SL, de Waard JR (2003) Biological identifications through DNA barcodes. Proc R Soc B 270(1512):313–321
Heethoff M, Domes K, Laumann M, Maraun M, Norton RA, Scheu S (2007) High genetic divergences indicate ancient separation of parthenogenetic lineages of the oribatid mite Platynothrus peltifer (Acari, Oribatida). J Evol Biol 20:392–402
Hewitt GM (2004) Genetic consequences of climatic oscillations in the Quaternary. Philos Trans R Soc Lond B Biol Sci 359(1442):183–195
Hoang DT, Chernomor O, von Haeseler A, Minh BQ, Vinh LS (2018) UFBoot2: improving the ultrafast bootstrap approximation. Mol Biol Evol 35(2):518–522
Kalyaanamoorthy S, Minh BQ, Wong TKW, von Haeseler A, Jermiin LS (2017) ModelFinder: fast model selection for accurate phylogenetic estimates. Nat Methods 14:587–589
Korstian KM, Hale AM, Williams DA (2015) Genetic diversity, historic population size, and population structure in 2 North American tree bats. J Mamm 96(5):972–980
Kováč Ľ, Parimuchová A, Miklisová D (2016) Distributional patterns of cave Collembola (Hexapoda) in association with habitat conditions, geography and subterranean refugia in the Western Carpathians. Biol J Linn Soc 119(3):571–592
Kreipe V, Corral-Hernández E, Scheu S, Schaefer I, Maraum M (2015) Phylogeny and species delineation in European species of the genus Steganacarus (Acari, Oribatida) using mitochondrial and nuclear markers. Exp Appl Acarol 66(2):173–186
Krivolutsky DA, Lebedeva NV (2004) Oribatid mites (Oribatei, Acariformes) in bird feathers: non-passerines. Acta Zool Litu 14(1):26–45
Kumar S, Stecher G, Li M, Knyaz C, Tamura K (2018) MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol 35(6):1547–1549
Laumann M, Norton RA, Weigmann G, Scheu S, Maraun M, Heethoff M (2007) Speciation in the parthenogenetic oribatid mite genus Tectocepheus (Acari, Oribatida) as indicated by molecular phylogeny. Pedobiologia 51(2):111–122
Lebedeva NV, Krivolutsky DA (2003) Birds spread soil microarthropods to arctic islands. Dokl Biol Sci 391:329–332
Lehmitz R, Decker P (2017) The nuclear 28S gene fragment D3 as species marker in oribatid mites (Acari, Oribatida) from German peatlands. Exp Appl Acarol 71(3):259–276
Lehmitz R, Russel D, Hohberg K, Christian A (2011) Wind dispersal of oribatid mites as mode of migration. Pedobiologia 54(3):201–207
Lehmitz R, Russell D, Hohberg K, Christian A, Xylander WER (2012) Active dispersal of oribatid mites into young soils. Appl Soil Ecol 55:10–19
Leigh JW, Bryant D (2015) PopART: full-feature software for haplotype network construction. Methods Ecol Evol 6(9):1110–1116
Librado P, Rozas J (2009) DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25:1451–1452
Ľuptáčik P (2004) Príspevok k rozšíreniu Gemmazetes cavaticus (Kunst, 1962) (Acarina, Oribatida, Thyrisomidae) v Európe so zameraním na územie Slovenska. Book of Abstracts Zoologické dny. Brno, Brno
Ľuptáčik P, Miko L (2003) Oribatid mites (Acarina, Oribatida) of Slovak caves. Subterr Biol 1:25–29
Luxton M (1972) Studies on the oribatid mites of a Danish beech wood soil. I. Nutritional biology. Pedobiologia 12(3):434–463
Maraun M, Scheu S (2000) The structure of oribatid mite communities (Acari, Oribatida): patterns, mechanisms and implications for future research. Ecography 23:374–383
Maraun M, Heethoff M, Scheu S, Norton RA, Weigmann G, Thomas RH (2003) Radiation in sexual and parthenogenetic oribatid mites (Oribatida, Acari) as indicated by genetic divergence of closely related species. Exp Appl Acarol 29(3–4):265–277
Marshall VG (1972) Comparison of two methods of estimating efficiency of funnel extractors for soil microarthropods. Soil Biol Biochem 4:417–426
Nguyen LT, Schmidt HA, von Haeseler A, Bui QM (2015) IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol 32(4):268–274
Norton RA, Bonamo PM, Grierson JD, Shears WA (1988) Oribatid mite fossils from a terrestrial Devonian deposit near Gilboa, New York. J Paleontol 62:259–269
Ojala R, Huhta V (2001) Dispersal of microarthropods in forest soil. Pedobiologia 45:443–450
Pachl P, Lindl AC, Krause A, Scheu S, Schaefer I, Maraun M (2017) The tropics as an ancient cradle of oribatid mite diversity. Acarologia 57(2):309–322
Papadopoulou A, Anastasiou I, Keskin B, Vogler AP (2009) Comparative phylogeography of tenebrionid beetles in the Aegean archipelago: the effect of dispersal ability and habitat preference. Mol Ecol 18(11):2503–2517
Parimuchová A, Kováč Ľ, Žurovcová M, Miklisová D, Paučulová L (2017) A glacial relict in the Carpathian caves—population variability or a species complex? Arthropod Syst Phylo 75(3):351–362
Pfingstl T, Baumann J, Lienhard A (2019) The Caribbean enigma: the presence of unusual cryptic diversity in intertidal mites (Arachnida, Acari, Oribatida). Org Divers Evol 19(4):609–623
Rambaut A, Drummond AJ, Xie D, Baele G, Suchard MA (2018) Posterior summarisation in Bayesian phylogenetics using tracer 1.7. Syst Biol 67(5):901–904
Retallack GJ (2001) Cenozoic expansion of grasslands and climatic cooling. J Geol 109(4):407–426
Ronquist F, Teslenko M, van der Mark P, Ayres DL, Darling A, Höhna S, Larget B, Liu L, Suchard MA, Huelsenbeck JP (2012) MRBAYES 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol 61(3):539–542
Rosenberger MJ (2010) Phylogeography in sexual and parthenogenetic European Oribatida. Dissertation, Göttingen Centre for Biodiversity and Ecology
Rosenberger MJ, Maraun M, Scheu S, Schaefer I (2013) Pre- and post-glacial diversifications shape genetic complexity of soil-living microarthropod species. Pedobiologia 56:79–87
Schaefer I, Norton RA, Scheu S, Maraun M (2010) Arthropod colonization of land-linking molecules and fossils in oribatid mites (Acari, Oribatida). Mol Phylogenet Evol 55:113–121
Schäffer S, Pfingstl T, Koblmüller S, Winkler KA, Sturmbauer Ch, Krisper G (2010) Phylogenetic analysis of European Scutovertex mites (Acari, Oribatida, Scutoverticidae) reveals paraphyly and cryptic diversity: a molecular genetics and morphological approach. Mol Phyl Evol 55:677–688
Schäffer S, Kerschbaumer M, Koblmüller S (2019) Multiple new species: cryptic diversity in the widespread mite species Cymberemaeus cymba (Oribatida, Cymbaeremaeidae). Mol Phylogenet Evol 135:185–192
Schuppenhauer MM, Lehmitz R, Xylander WER (2019) Slow-moving soil organisms on a water highway: aquatic dispersal and survival potential of Oribatida and Collembola in running water. Mov Ecol. https://doi.org/10.1186/s40462-019-0165-5
Skoracka A, Magalhães S, Rector BG, Kuczyński L (2015) Cryptic speciation in the Acari: a function of species lifestyles or our ability to separate species? Exp Appl Acarol 67(2):165–182
Starý J (2008) Diversity and distribution of oribatid mites (Acari: Oribatida) in caves of Czech Republic. Acta Carsol Slovaka 46(1):185–197
Subías LS (2020) Listado sistemático, sinonímico y biogeográfico de los Ácaros Oribátidos (Acariformes: Oribatida) del mundo (excepto fósiles), 15a actualización. http://bba.bioucm.es/cont/docs/RO_1.pdf. Accessed June 2020
Tajima F (1989) Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. G3 (Bethesda) 123:585–595
Villesen P (2007) FaBox: an online toolbox for fasta sequences. Mol Ecol Notes 7(6):965–968
von Saltzwedel H, Mark M, Scheu S, Schaefer I (2014) Evidence for frozen niche variation in a cosmopolitan parthenogenetic soil mite species (Acari, Oribatida). PLoS ONE 9(11):e113268
von Saltzwedel H, Scheu S, Schaefer I (2016) Founder events and pre-glacial divergences shape the genetic structure of European Collembola species. BMC Evol Biol 16:148. https://doi.org/10.1186/s12862-016-0719-8
von Saltzwedel H, Scheu S, Schaefer I (2017) Genetic structure and distribution of Parisotoma notabilis (Collembola) in Europe: cryptic diversity, split of lineages and colonization patterns. PLoS ONE 12(2):e0170909
Waters JM, Fraser CI, Hewitt GM (2013) Founder takes all: density-dependent processes structure biodiversity. Trends Ecol Evol 28(2):78–85
Weigmann G (2006) Die Tierwelt Deutschlands, Teil 76: Hornmilben (Oribatida). Goecke and Evers, Keltern
Xia X (2017) DAMBE6: new tools for microbial genomics, phylogenetics and molecular evolution. J Hered 108:431–437
Xia X, Xie Z, Salemi M, Chen L, Wang Y (2003) An index of substitution saturation and its application. Mol Phylogenet Evol 26:1–7
Yosii R (1956) Monographie zur Höhlencollembolen Japans, vol 3. Biological Laboratory, Kyoto University, Kyoto, p 1
Żbikowska-Zdun K, Piksa K, Smaczyńska A (2009) Variation of selected morphological characters of the cave mite Oribella cavatica Kunst, 1962 (Acari, Oribatida). Biol Lett 46(2):123–127
Acknowledgements
The research was supported by the Czech Academy of Sciences (under Research Plan No. AV0Z606960521), by two grant projects of the Czech Science Foundation (projects No. P504/12/1218 and No. 14-09231S) and one project of the Slovak Research and Development Agency (APVV-17-0477). The research in Czech caves was undertaken in the collaboration with the Cave Administration of Czech Republic. We want to thank to Dr. Koudelka (Head of the Javoříčské Caves) for cooperation and enabling the access to the Javoříčské Caves; Dr. Štefka (Head of the Moravian Karst PLA), Dr. Tůma and Dr. Kovařík for cooperation and help with the field sampling in Sloupsko-šošůvské Caves and Amatérská Cave. We thank also to the Slovak Caves Administration and to the Administrations of Šumava NP, Wigry NP, Tatranský NP and Slovenský kras NP for their permission to conduct the study in the national parks.
Funding
This study was supported by the Academy of Sciences of the Czech Republic (under Research Plan No. AV0Z606960521), by two grant projects of the Czech Science Foundation (projects No. P504/12/1218 and No.14-09231S) and by one project of the Slovak Research and Development Agency (APVV-17-0477).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest
The authors have no conflict of interest to declare that are relevant to the content of this article.
Ethics approval
No approval of research ethics committees was required to accomplish the goals of this study because experimental work was conducted with an unregulated invertebrate species.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
About this article

Cite this article
Kokořová, P., Žurovcová, M., Ľuptáčik, P. et al. Distinct phylogeographic patterns in populations of two oribatid mite species from the genus Pantelozetes (Acari, Oribatida, Thyrisomidae) in Central Europe. Exp Appl Acarol 83, 493–511 (2021). https://doi.org/10.1007/s10493-021-00605-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10493-021-00605-7