
Abstract
Actinomycetes are antibiotic-producing filamentous bacteria that have a mycelial life style. The members of the three genera classified in the family Streptomycetaceae, namely Kitasatospora, Streptacidiphilus and Streptomyces, are difficult to distinguish using phenotypic properties. Here we present biochemical and genetic evidence that helps underpin the case for the continued recognition of the genus Kitasatospora and for the delineation of additional Kitasatospora species. Two novel Kitasatospora strains, isolates MBT63 and MBT66, and their genome sequences are presented. The cell wall of the Kitasatospora strains contain a mixture of meso-and LL-diaminopimelic acid (A2pm), whereby a single DapF surprisingly suffices to incorporate both components into the Kitasatospora cell wall. The availability of two new Kitasatospora genome sequences in addition to that of the previously sequenced Kitasatospora setae KM-6054T allows better phylogenetic comparison between kitasatosporae and streptomycetes. This showed that the developmental regulator BldB and the actin-like protein Mbl are absent from kitasatosporae, while the cell division activator SsgA and its transcriptional activator SsgR have been lost from some Kitasatospora species, strongly suggesting that Kitasatospora have evolved different ways to control specific steps in their development. We also show that the tetracycline-producing strain “Streptomyces viridifaciens” DSM 40239 not only has properties consistent with its classification in the genus Kitasatospora but also merits species status within this taxon.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The phylum Actinobacteria (Goodfellow 2012) includes many morphologically complex mycelium-forming bacteria that are of great importance for biotechnology due to their ability to produce a large array of natural products, including antibiotics, anticancer agents and immunosuppressants (Hopwood 2007). The rapid spread of multiple drug resistant pathogens and the discovery that whole genomes of filamentous actinomycetes are rich in biosynthetic gene clusters accounts for the renewed research focus on these organisms (Baltz 2008). The revolution caused by next-generation sequencing technologies (Shendure and Lieberman Aiden 2012) has greatly accelerated the release of new actinomycete genome sequences. Thus, as understanding of the genetics of filamentous actinomycetes increases a large number of biosynthetic gene clusters are being unveiled, offering new options for genome mining-based drug discovery. However, new challenges are posed by the need to find new ways to activate gene clusters so as to identify the products they specify (Baltz 2008; Craney et al. 2012; Zhu et al. 2014a), and by the requirement for rapid and sound classification of putatively novel actinomycete species. The wealth of genome data gives more detailed insights into the evolution of genes, identifying classes of proteins that are specific to organisms at generic and family ranks (Kirby 2011a), while the distribution of genes provides valuable insights into the role of proteins or protein families in cellular processes, such as in development and secondary metabolism.
Many biotechnologically important actinomycetes classified in the family Streptomycetaceae, have a complex mycelial life cycle and are a striking example of multicellular bacteria (Claessen et al. 2014; Flärdh and Buttner 2009). Their life cycle starts with a single spore that germinates to form young vegetative hyphae, which then grow out following a process of hyphal growth and branching to produce a branched vegetative (substrate) mycelium (Chater and Losick 1997). During the reproductive phase aerial hyphae differentiate into long chains of spores following a complex cell division event whereby ladders of septa are produced within a short time span (Jakimowicz and van Wezel 2012; McCormick 2009). Genes that control the onset of morphological differentiation are referred to as bld (bald) genes (Merrick 1976), and those that control the differentiation of aerial hyphae into mature spores as whi (white) genes, indicating the lack of production of the grey spore pigment (Chater 1972).
Several families of developmental regulatory proteins occur exclusively in filamentous actinomycetes, such as the WhiB-like proteins (Wbl; (Soliveri et al. 2000)) and SsgA-like proteins (SALPs), which reflects the specific demands set by the complex sporulation programme. The SALPs are cell division regulators that occur exclusively in morphologically complex actinomycetes (Jakimowicz and van Wezel 2012; Traag and van Wezel 2008), with a suggestive linkage between the number of SALP paralogues and the complexity of the developmental process; as a rule of thumb, actinomycetes which produce single spores have a single SALP, which invariably is an orthologue of SsgB (Xu et al. 2009), those that form short spore chains two SALPs, while actinomycetes with complex development, such as Frankia and Streptomyces species, typically have multiple SALPs. The model organisms Streptomyces coelicolor (Bentley et al. 2002) and Streptomyces lividans (Cruz-Morales et al. 2013), bona fide members of Streptomyces violaceoruber, contain seven SALPs (SsgA-G). All sporulating actinomycetes contain SsgB, which is essential for sporulation (Keijser et al. 2003) and recruits the cell division protein FtsZ to future septum sites (Willemse et al. 2011).
The extraordinary evolution, in particular of the SsgB protein, with (almost) complete conservation of its protein sequence within a particular genus (maximum of one amino acid [aa] change), and large divergence even between related genera, led to a proposed new taxonomic tool for the classification of morphologically complex actinomycetes (Girard et al. 2013). This morpho-taxonomic approach complements the more traditional molecular analyses based on the divergence of 16S rRNA and rpoB genes, and is important as the latter do not always provide sufficient resolution when comparing closely related species nor do they confidently discriminate between or unite sister genera within families.
The taxonomic status of the genus Kitasatospora (Omura et al. 1982) within the family Streptomycetaceae has been a matter of dispute for many years (Ludwig et al. 2012). The genus was reduced to a synonym of the genus Streptomyces by Wellington et al. (1992), and then re-established as a separate genus by Zhang et al. (1997). The status of the genus is still a matter of debate (Kämpfer 2012; Labeda et al. 2012), though relationships based on the phylogeny of the SsgA-like proteins data suggest that Kitasatospora should probably be seen as a sister genus to Streptomyces within the family Streptomycetaceae (Girard et al. 2013). Genome sequence comparison is an important means of phylogenetic comparison. Until now, the only available Kitasatospora genome sequence was that of Kitasatospora setae KM-6054T, the type strain of the genus (Ichikawa et al. 2010). Indeed, Ishikawa and colleagues noted that many of the genes related to morphological differentiation in Streptomyces were highly conserved in the K. setae strain though there were some differences as exemplified by the apparent absence of the Amfs (SapB) class of surfactant proteins and differences in the copy number and variations of paralogous components involved in cell wall synthesis. In addition, multilocus phylogenetic analysis based on amino acid sequences clearly placed the K. setae strain outside the genus Streptomyces.
In this study, we present new insights into the evolution and origin of Kitasatospora and provide further phylogenetic evidence that this taxon should retain its generic status within the family Streptomycetaceae. We present the draft genome sequences of Kitasatospora strains MBT63 and MBT66, which were isolated from a Himalayan soil, and show that genes related to morphological differentiation and cell division have been lost in kitasatosporae, including bldB, which is required for the formation of aerial mycelia in streptomycetes (Pope et al. 1998), the sporulation gene whiJ (Ainsa et al. 2010) and the gene for the cytoskeletal protein Mbl (Soufo and Graumann 2003), while ssgA and its transcriptional activator gene ssgR (Traag et al. 2004) are apparently in the process of being lost from kitasatosporae. We also provide preliminary, but compelling evidence that the two Kitasatospora isolates and the invalidly described “Streptomyces viridifaciens” strain DSM 40239 form new centers of taxonomic variation within the genus Kitasatospora that merit recognition as new species.
Results and discussion
Isolation, classification and whole-genome sequencing of Kitasatospora strains MBT63 and MBT66
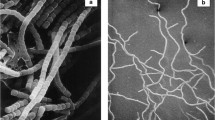
These strains were isolated from a Himalayan soil sample and provisionally assigned to the genus Kitasatospora on the basis of partial 16S ribosomal RNA analysis (Zhu et al. 2014b). This assignment was underpinned in the present study as it was shown that whole-organism hydrolysates of their substrate mycelia contained meso-diaminopimelic acid (meso-A2pm). When grown on SFM agar plates strain MBT66 secretes a brown diffusible pigment and its aerial hyphae differentiate into chains of exospores (Fig. S1). Detailed analysis of the spore chains by cryo-scanning electron microscopy revealed chains that were longer than those produced by Kitasatospora strain MBT63, which resembled those formed by the model organism S. coelicolor A3(2) (Fig. 1). To gain more insight into the evolution of Kitasatospora strains, and the genes involved in their morphological development, a draft genome sequence was generated for strain MBT66 by Illumina paired-end sequencing combined to Pacific Biosciences reads, which were assembled into 45 scaffolds (Table 1). A less detailed draft genome sequence was generated for Kitasatospora strain MBT63 by Illumina technology, which yielded 849 contigs. The genome of Kitasatospora strain MBT63 has a G + C content of 73.0 %, a genome size of 9.9 Mb and encodes a predicted 8,651 proteins, while that of Kitasatospora strain MBT66 has a G + C content of 73.2 %, a genome size of 10.4 Mb and encodes a predicted 8,827 proteins (Table 1). With 8,783,278 bp (8.8 Mb) and encoding a predicted 7,569 proteins, the genome of K. setae KM-6054T is significantly smaller (Ichikawa et al. 2010). Putative proteins and RNAs encoded by the genomes were annotated as described in the Materials and Methods section (Fig. S2 and Table 1). No major differences were observed for gene function predictions by RAST between the Himalayan Kitasatospora strains and K. setae KM-6054T (Fig. S2).

Scanning electron micrographs of Kitasatospora strains MBT63 and MBT66 grown on SFM agar plates for 5 days at 30 °C. Bar, 2 μm
The chromosomes of Streptomyces, unlike those of most other bacteria, are linear, with the telomeres containing inverted repeats that are covalently bound by terminal proteins (Yang et al. 2002; Lin et al. 1993). The presence of homologues of the tapA (SCO7733 in S. coelicolor), tpgA (SCO7734) and ttrA (SCO0002 and SCO7845) genes is an obvious marker for linearity of the chromosome (Kirby et al. 2008), and identification of these genes in K. setae supports the notion that its genome is also linear (Ichikawa et al. 2010). Analysis of the genomes of Kitasatospora strains MBT63 and MBT66 revealed the presence of orthologues of ttrA, tapA and tpgA, with the latter two immediately adjacent to one another, suggesting they are true functional orthologues. There is between 42 and 49 % aa identity between the predicted TapA and TpgA proteins of S. coelicolor and the Kitasatospora strains. The homology between the putative ttrA1 and ttrA2 genes of S. coelicolor A3(2) and of the three Kitasatosporae is even lower (i.e. lower than 40 % aa identity between the predicted gene products). Considering this low conservation and the lack of information concerning the precise location of the chromosomal ends in the draft MBT genomes, further and more detailed characterization is needed to ascertain the linearity of the genomes of Kitasatospora MBT63 and MBT66.
Molecular taxonomy underscores the generic status of Kitasatospora
The classification of the Kitasatospora strains based on 16S rRNA gene sequences and RpoB and RecA protein sequences are shown in Figs. S3, S4 and S5, respectively. It is apparent from the 16S rRNA tree (Fig. S3) that Kitasatospora strains MBT63 and MBT66 cluster with the type strains of validly published Kitasatospora species albeit within the evolutionary radiation occupied by type strains of Streptomyces species, a result supported by corresponding RpoB and RecA sequences. Additional genotypic and phenotypic data are needed to underpin the generic status of Kitasatospora. We recently showed the value of SsgA-like protein sequences (SALPs) as additional molecular taxonomic markers for sporulating actinomycetes (Girard et al. 2013). The conservation of SsgB sequences—namely identical or nearly identical within a genus and at the same time highly variable even between related genera—allows accurate classification of actinomycetes at the level of the genus. The SsgB-based phylogenetic tree (Fig. S6) shows that Kitasatospora strains MBT63 and MBT66 map with the type strain of K. setae and away from the Streptomyces strains. With three to four aa changes between the SsgB orthologues from the Kitasatospora and Streptomyces strains, as opposed to complete identity or one aa change between the type strains of Streptomyces, the differences are small but significant. Moreover, at the nucleotide (nt) level the conservation is also much higher between K. setae KM-6054T and Kitasatospora strains MBT63 and MBT66 when compared to the Streptomyces ssgB sequences (Table S1), thereby providing further grounds for the continued recognition of the genus Kitasatospora.
A second SALP of value in the classification of morphologically complex actinomycetes is SsgA, the aa sequence of which is a reliable predictive marker for the ability of streptomycetes to produce spores in submerged culture (Girard et al. 2013). The SsgA homologue of Kitasatospora strain MBT63 (on contig 282) only shares 50–55 % aa identity with SsgA from streptomycetes (53 % aa identity with S. coelicolor SsgA (SCO3926)), and is closer to the SsgA orthologue of K. setae KM-6054T (63 % aa identity) (Fig. S7). However, gene synteny evidence identified the gene as a true ssgA (Fig. 2). Six signature aa residues were identified in SsgA from streptomycetes that together serve as markers for the LSp (liquid culture sporulation) or NLSp (no liquid culture sporulation) branches of the streptomycetes (Girard et al. 2013). However, this conservation is lost in the Kitasatospora strains as neither signature can be recognised.

Genomic region around ssgRA in Kitasatospora setae KM-6054T and S. coelicolor A3(2), and the corresponding regions in Kitasatospora strains MBT63 and MBT66. Genes in the MBT strains are named after the locus of their corresponding orthologue in K. setae. Color codes refer to direct orthologues with S. coelicolor (white: no direct orthologue found in S. coelicolor)
The draft genome sequence of Kitasatospora strain MBT66 was further scaffolded by alignment to the reference genomes of S. coelicolor A3(2) and K. setae KM-6054T using the R2CAT program (see Materials and Methods). Subsequent analysis by genome alignment using MAUVE further supported the notion that the genome of Kitasatospora strain MBT66 is more closely related to the K. setae strain than to S. coelicolor A3(2). Indeed, syntenous regions were significantly larger when the genome of Kitasatospora strain MBT66 was compared to that of K. setae KM-6054T (Fig. S8a) than to the S. coelicolor genome (Fig. S8b). MAUVE analysis revealed the lowest conservation at the extremities of the genomes, as described previously (Kirby 2011a). Thus, the phylogenetic trees together with the divergence of the different phylogenetic markers and the whole genome comparison strongly support the view that the genus Kitasatospora is distinct from the genus Streptomyces; they also show that Kitasatospora strains MBT63 and MBT66 represent putative novel species of Kitasatospora.
A single DapF suffices for incorporation of meso-and LL-diaminopimelic acid into the spore wall of Kitasatospora strains MBT63 and MBT66
Kitasatospora and Streptomyces strains are very difficult to distinguish based on phenotypic characteristics, including morphological criteria, but can be separated on the basis of cell wall composition (Kämpfer 2012). Thus, whole-organism hydrolysates of streptomycetes are rich in LL-diaminopimelic acid (A2pm) whereas the substrate mycelium of kitasatosporae contain meso-A2pm, while spores contain the corresponding LL-isomer. DapF is responsible for the isomerisation of LL-A2pm into meso-A2pm, whereas MurE incorporates both types of A2pm into peptidoglycan ((Ichikawa et al. 2010) and references therein). Three dapF paralogues occur in K. setae KM-6054T, while only a single dapF gene is found in most streptomycetes. KSE_32600 and KSE_53750 are closely related to the dapF found in all Streptomyces species whereas KSE_32630 is closer to dapF orthologues found in bacteria that contain meso-A2pm. It has been suggested that differential regulation of the three dapF paralogues in the K. setae strain is responsible for the changes in A2pm composition during development (Ichikawa et al. 2010).
As anticipated, a single murE was found in Kitasatospora strains MBT63 and MBT66, as in K. setae KM-6054T. However, only one dapF gene was identified in strains MBT63 and MBT66, in both cases a direct orthologue of KSE_53750 (79 % aa identity between the predicted gene products and conserved gene synteny; not shown). Phylogenetic analysis indicated that the DapF orthologues of Kitasatospora strains MBT63 and MBT66 are closest to the “LL-A2pm branch” of DapF homologues (Fig. 3). Interestingly, analysis of the cell-wall composition of the spores of Kitasatospora strains MBT63 and MBT66 revealed that, as for the K. setae strain, the hyphae of these organisms contained a mixture of the isomers meso-2,6-diaminopimelate (meso-A2 pm) and LL-2,6-diaminopimelate (LL-A2 pm), while the spores contained only LL- A2pm. This strongly suggests that a single DapF orthologue is sufficient to direct the differential incorporation of both the meso-and LL-isomers of A2pm into the cell wall through various stages of development in Kitasatospora strains. Within the streptomycetes, Streptomyces avermitilis MA-4680T is exceptional as it has two DapF paralogues, as opposed to the one routinely found in streptomycetes, whereby one (SAV_3161) does not cluster in the LL-A2pm branch (Fig. 3).

Maximum-likelihood phylogenetic tree for DapF protein sequences in different actinomycetes. The type of A2pm (diaminopimelic acid) found in the cell walls of the various actinomycetes is indicated. For input sequences see Supplemental Data file 1
BldB, Mbl and WhiJ are absent in Kitasatospora
Close inspection of the genes that play a major role in the control of Streptomyces development and cell division shows that many genes are conserved between them, including genes in the dcw (division and cell wall) cluster that includes the fts (cell division) and mur (cell wall biosynthesis) genes (Fig. S9), and nearly all of the bld and whi developmental regulatory genes. In contrast, bldB and whiJ are absent from K. setae KM-6054T and Kitasatospora strains MBT63 and MBT66. Considering the very close relationship between Kitasatospora and Streptomyces strains, this supports the hypothesis that bldB and whiJ may be Streptomyces-specific genes (Kirby et al. 2011b). However, since whiJ is also absent from a number of streptomycetes, this gene does not qualify as a suitable marker. It was mentioned previously that ramS/amfS, which specify the lantibiotic-like developmental signalling protein SapB (Kodani et al. 2004; Willey et al. 1991), is also absent from K. setae (Ichikawa et al. 2010), but considering the presence of several genes for LanA precursors and corresponding LanBC/LanM type modifying enzymes in kitasatosporae (see below), the absence of SapB in this genus still requires experimental validation.
BldB is a small protein that is required for the initiation of development and antibiotic production, but it also plays a role in carbon catabolite repression that has not yet been resolved (Pope et al. 1998). The nearest orthologue to BldB is KSE_16220, but this has its highest similarity to SCO7246; there is 60–67 % aa identity between the predicted gene products, as opposed to 45–53 % aa identity to BldB, and gene synteny confirms that it is indeed a direct orthologue of SCO7246 (not shown). Streptomycetes have an unusually extensive cytoskeleton, which plays a role in among other cell-wall stability, apical growth, and development (Celler et al. 2013). The cytoskeletal genes identified in streptomycetes are largely conserved in Kitasatospora, including scy (Holmes et al. 2013) and filP (Bagchi et al. 2008) for intermediate filament-like proteins, but mbl for the actin-like protein Mbl, which helps ensuring cell-wall integrity and cell shape in streptomycetes (Heichlinger et al. 2011), is missing from the Kitasatospora genomes. The absence of bldB is particularly intriguing. As mentioned above, Kitasatospora and Streptomyces are difficult to distinguish phenotypically and the nearly complete conservation of the bld and whi genes in these genera strongly suggests a highly similar developmental control network. Since BldB is required for development of streptomycetes (Eccleston et al. 2002), its absence in Kitasatospora strains could provide new insights into the function of this gene, e.g. by analysing how the bldB developmental checkpoint is by-passed in Kitasatospora. It should be noted that the requirement of many of the bld genes for differentiation is medium-dependent in streptomycetes, with a developmental block on glucose-containing media, while growth on a non-repressing carbon source such as mannitol often allows the colonies to by-pass the developmental block and enter aerial growth (Pope et al. 1996). In line with the notion that carbon catabolite repression (CCR) may be a dominant factor, deletion of the glkA gene for glucose kinase, which controls CCR in streptomycetes (Angell et al. 1994), allows many bld mutants to differentiate on glucose-containing media (van Wezel and McDowall 2011). Therefore, perhaps the lack of BldB reflects a different connection between nutrient utilization and development in Kitasatospora (and other actinomycetes).
Interestingly, the bldB gene appears to be deleted with what may be described as surgical precision from the Kitasatospora genomes, with all flanking genes present-though not in identical gene order-as compared to S. coelicolor A3(2) (Fig. 4). This is even more surprising when one realises that the five genes that lie upstream of bldB in S. coelicolor (SCO5724-SCO5728) have been lost from the S. lividans genome, while an insertion of 11 genes occurred downstream of bldB (Cruz-Morales et al. 2013; Lewis et al. 2010). In other words, as compared to S. coelicolor, Kitasatospora has maintained all genes except bldB, while in S. lividans many genes around bldB have been lost, and others sustained rearrangements (Fig. 4).

Genomic region around bldB (SCO5723, in red) of the S. coelicolor A3(2) genome and the corresponding region in S. lividans 66 and in three Kitasatospora strains, whereby the genome organization is identical in K. setae and Kitasatospora strains MBT63 and MBT66. Note that apart from some rearrangements, only bldB is missing in the kitasatosporae, while conversely, bldB is conserved in S. lividans while many of the flanking genes have been lost or moved. The Kitasatospora genes are labelled according to the K. setae nomenclature. Colour codes refer to direct orthologues with S. coelicolor A3(2)
Loss of ssgRA from Kitasatospora strains
In terms of the SALPs, five (SsgA, SsgB, SsgD, SsgE and SsgG) occur in nearly all streptomycetes, while a few additional strain-specific SALPs are often found. Kitasatospora setae KM-6054T contains twelve SALPs while Kitasatospora strains MBT63 and MBT66 likely have eight and seven SALPs, respectively. Phylogenetic linkage between the Kitasatospora SALPs and those from several model streptomycetes is presented (Fig. 5). Most SALPs of Kitasatospora strains MBT63 and MBT66 cluster with those from the K. setae strain as opposed to those of Streptomyces species. Of the conserved ssg genes, likely orthologues of ssgA, ssgB, ssgD and ssgG were identified in the Kitasatospora strains and their presence further validated by gene synteny evidence (Fig. S10 and S11). For the ssgD orthologues in the Kitasatospora strains the gene synteny was less convincing, with only one neighbouring gene (the homologue of SCO6726 in S. coelicolor A3(2)) found in the vicinity of ssgD (SCO6722) in Kitasatospora strain MBT66 and in K. setae KM-6054T, while it was absent in Kitasatospora strain MBT63. However, the three hypothetical ssgD homologues found in the kitasatosporae convincingly cluster with the ssgD sequences of streptomycetes (Fig. 5).

Maximum-likelihood phylogenetic tree of SALP protein sequences in Kitasatospora setae and Kitasatospora strains MBT63 and MBT66. The asterisks indicate SALPs for which synteny was analyzed in this study. For input sequences see supplemental data file 2
SsgA has not been identified in any actinobacterial family outside the family Streptomycetaceae. It has been found in all sequenced Streptomyces species while an ssgA orthologue is present in K. setae KM-6054T, together with its activator gene ssgR, which encodes an IclR-family transcriptional regulator that activates the transcription of ssgA (Traag et al. 2004). Surprisingly, neither ssgA nor ssgR were found in Kitasatospora strain MBT66, while only ssgA was found in Kitasatospora strain MBT63 even though most of the flanking genes are still present. The absence of the sporulation activator gene ssgA in MBT66 may explain its poor sporulation (see Fig. 1). There are homologues to some of the neighbouring genes (SCO3924, SCO3922 and SCO3918), but the gene order is shuffled (Fig. 2). Kitasatospora strain MBT66 does not possess an ssgA homologue anywhere else in the genome. It would be interesting to see whether re-introduction of ssgA would restore normal development to this strain. So far, however, we have not been able to obtain transformants with plasmids routinely used for genetic manipulation of streptomycetes. The absence of ssgR in Kitasatospora strain MBT63 and of ssgRA in Kitasatospora strain MBT66, was corroborated by PCR analysis and resequencing (data not shown). This strongly suggests that these genes, which activate sporulation-specific cell division, and play an important role in germination and mycelial morphology in Streptomyces (Kawamoto et al. 1997; Noens et al. 2007; van Wezel et al. 2006), are being lost from members of the genus Kitasatospora. It is not clear how Kitasatospora by-passes the important sporulation control proteins BldB, WhiJ and SsgR/SsgA given that all other important sporulation genes found in streptomycetes are highly conserved in kitasatosporae.
Gene clusters for natural products
For further comparison of the three Kitasatospora genomes, scaffolded MBT63 and MBT66 contigs and the K. setae KM-6054T genome sequence were aligned to one another using MAUVE. As expected, synteny blocks are larger and show higher nucleotide conservation in the central core region which contains most of the highly conserved household genes, while the chromosomal ends contain more variable genes, such as those involved in natural products biosynthesis (Bentley et al. 2002; Kirby 2011a). Two small neighboring synteny blocks are striking as they are highly conserved in the genomes of Kitasatospora strains MBT63 and MBT66, but absent in that of the K. setae strain (Fig. 6a). Closer inspection revealed that this genomic region contains a lantibiotic type I biosynthesis cluster (Fig. 6b). Lantibiotics are compounds with antibiotic activity and little development of resistance in the target organisms (Willey and van der Donk 2007). These compounds are ideally suited for discovery by genome mining, as the ribosomally synthesized gene product can be predicted directly from the primary sequence (Foulston and Bibb 2010). The region found in the genomes of Kitasatospora strains MBT63 and MBT66 contain the classical type I elements: two biosynthetic genes (lanB and lanC), and a lanA gene product that contains a conserved FDLD motif in the leader peptide (Fig. 6b). These genes are very highly conserved between the two Himalayan genomes (90–100 % identity at nt level). This high level of identity and the presence of prophage proteins (50 kb upstream of the cluster) and CRISPR genes in both strains, as well as some transposase and integrase genes in Kitasatospora strain MBT66, suggests that this cluster results from horizontal transfer.

Genome comparison for Kitasatospora strains a Mauve alignment of Kitasatospora setae KM-6054T genome (top) to MBT63 (middle) and MBT66 (bottom) R2CAT-scaffolded contigs. Each block outlines a region of the genome sequence that aligns to part of another genome, and is presumably homologous and internally free from genomic rearrangements. Blocks above the central line indicate forward orientation relative to the first genome sequence, blocks indicate regions that align in the reverse complement orientation. Inside each block, the height of the similarity profile corresponds to the average level of conservation in that region of the genome sequence. The red and brown blocks marked by an asterisk are conserved in Kitasatospora strains MBT63 and MBT66 but are absent in K. setae KM-6054T b Part of the lantibiotic cluster (in contig 0960 of Kitasatospora strain MBT63 and scaffold 5 of Kitasatospora strain MBT66) found in the region marked by the asterisk in panel (A). Colors indicate putative functions as follows: transporter (purple), regulator (orange), lantibiotic dehydratase/cyclase (yellow) and lantibiotic precursor (pink). The predicted aa sequence of the LanA-type precursor peptide is shown, with the conserved FDLD motif in the leader peptide in red
Kitasatospora strains MBT63 and MBT66 were recently identified as promising antibiotic producers (Zhu et al. 2014b), and produce antibiotics that inhibit growth of a wide range of Gram-positive and Gram-negative multi-drug resistant pathogens. However, the antimicrobial compounds they produce have not yet been identified. Natural product gene clusters of Kitasatospora strain MBT66 were predicted using antiSMASH (Medema et al. 2011) and an in-house developed genome annotation pipeline (GG and GPvW, unpublished) (Table S2). In addition to the lantibiotic cluster described above, one more cluster was found to be common (although with lower conservation) between Kitasatospora strains MBT63 and MBT66. Lantibiotic genes lanABC are homologous to one another between MBT63 contig 0567 and MBT66 contig 0014, with 92 % identity at aa level for the LanA precursors. However, the flanking regions are different, including a phage integrase downstream of lanC in the genome of Kitasatospora strain MBT63. Kitasatospora strain MBT66 is further predicted to have two more lantibiotic clusters, one of which harbors a clear lanA. Other putative clusters for natural products in Kitasatospora strain MBT66 are summarised in Table S2 and can be visualised using the xhtml files given in the supplementary material.
In addition to the four lantibiotic clusters mentioned previously (two of which are conserved in K. setae KM-6054T, which has six in total), up to 20 NRPS/PKS clusters are predicted to be encoded by the Kitasatospora MBT66 genome (four of which are conserved in the K. setae strain), as well as gene clusters for a bacteriocin and a gamma-butyrolactone (conserved in K. setae KM-6054T, which has six in total), two for siderophores (conserved in K. setae), six terpene clusters (two conserved in K. setae) and three gene clusters of unknown type (none of which were found in K. setae). The partially assembled genome of Kitasatospora strain MBT63 is too fragmented for accurate prediction of natural product clusters. The K. setae genome is predicted by antiSMASH to harbour 11 gene clusters for NRPS/PKS, which is significantly less than found in Kitasatospora strain MBT66. A majority of the clusters found in the latter are not conserved in K. setae KM-6054T; the general pattern of conservation illustrates the high plasticity and a degree of horizontal transfer for this part of the genome.
New generic assignment for “Streptomyces viridifaciens” DSM 40239
We recently noticed that one of the strains in our collection, which was obtained from the DSMZ strain collection as Streptomyces viridifaciens DSM 40239 (ATCC 11989), contained an SsgB orthologue with a Kitasatospora signature (Fig. 7a). This strain, which produces tetracycline and chlortetracycline, was described as S. viridifaciens by Gourevitch and Lein (1955), but the name has not been validly published and hence has no formal standing in nomenclature. Whole-organism hydrolysates of the substrate mycelium and spores of the strain contained meso-A2pm and LL-A2pm, respectively providing further evidence that it should be classified in the genus Kitasatospora.

Phylogenetic analysis identifies “S. viridifaciens” as a likely member of the genus Kitasatospora. Maximum-likelihood phylogenetic trees with a limited number of species were prepared for a SsgB (protein sequence), b 16S rRNA (DNA sequence) and c SsgA (protein sequence). DapF was included in the phylogenetic tree presented in Fig. 3
To get better insight into the phylogeny of strain DSM 40239 and to further validate the suitability of SsgB as a phylogenetic marker, we analysed the 16S rRNA, dapF, ssgA and ssgR genes and also checked for the presence of bldB, mbl and whiJ. As anticipated, the 16S rRNA sequence clustered close to Kitasatospora (Fig. 7b). This outcome was confirmed by the fact that dapF clustered closely with the dapF of other kitasatosporae, strongly suggesting it is able to incorporate both meso-and LL-A2pm in its cell wall (Fig. 3), and by the absence of bldB, mbl and whiJ. “Streptomyces viridifaciens“DSM 40239 contains ssgA but not ssgR, and again ssgA belongs to the Kitasatospora branch of the phylogenetic tree (Fig. 7c). It is clear from these results that ‘S. viridifaciens’ DSM 40239 is not only an authentic member of the genus Kitasatospora, but merits recognition as a new putative species within this taxon though further comparative taxonomic studies are needed to confirm this. Screening the databases did not identify Kitasatospora-type SsgB proteins other than the ones discussed in this work, but it may be worthwhile to perform such a search in strain collections.
Concluding remarks
New genome sequences provide a source of much interesting and varied information: gene clusters for antibiotics, anticancer agents and other natural products as well as for industrial enzymes, but also new insights for fundamental science, especially evolution. In this work, the complete genome sequence of the putative novel Kitasatospora strain MBT66, supported by the draft sequence of Kitasatospora strain MBT63, provides an important wealth of new genomic data, new clues for the inferred evolution of the genera Kitasatospora and Streptomyces, and strong evidence that the former should retain its generic status. The evolution of developmental regulatory proteins is remarkable. The surprising absence of genes for the developmental proteins BldB and WhiJ and the actin-like cytoskeletal protein Mbl in all of the Kitasatospora strains, as well as the gradual loss in kitasatosporae of the genes for cell division activator SsgA and its transcriptional activator SsgR, suggest that the function of these proteins has been by-passed in Kitasatospora. It should be noted, however, that neither of these proteins is absolutely required for development under all growth conditions. Indeed, bldB mutants develop aerial hyphae and spores on mannitol-containing media, while whiJ and ssgA are rare examples of sporulation genes that are redundant under at least some growth conditions (Ainsa et al. 2010; van Wezel et al. 2000). These findings should lead to new insights with respect to the control of developmental processes in members of the family Streptomycetaceae. Furthermore, it provides biological support in addition to the strong phylogenetic evidence that kitasatosporae clearly merits generic status within the family Streptomycetaceae. It is also clear that”S. viridifaciens” DSM 40239 is a bona fide member of the genus Kitasatospora as it exhibits all of the taxonomic markers considered in this paper (in particular 16S rRNA, bldB, dapF, mbl and ssgRA). We believe that this may not be the only example; indeed, based on the fact that a number of streptomycetes rank closely to Kitasatospora in the 16S rRNA phylogenetic tree (Labeda et al. 2012), additional strains classified in the genus Streptomyces may turn out to belong to the genus Kitasatospora, such as for example Streptomyces purpeofuscus TSR6 (Genbank accession JF707864). The results of the present study suggest that the genus Kitasatospora is underspeciated. Insights into the evolution of development and natural product biosynthesis in morphologically complex actinomycetes will undoubtedly be further enhanced as many more genome sequences become available in the near future.
Materials and methods
Bacterial strains: source, growth, imaging and cell wall analysis
Kitasatospora strains MBT63 and MBT66 were isolated from Himalayan soil samples (Zhu et al. 2014b) and grown in liquid TSBS (tryptic soy broth with 10 % (w/v) sucrose) and on SFM (soy flour mannitol) agar plates at 30 °C. Strains were stored as frozen spores obtained from SFM-grown solid cultures. Imaging of strains was done by phase contrast microscopy, by stereo microscopy and by cryo-scanning electron microscopy, as described previously (Colson et al. 2008). “Streptomyces viridifaciens” DSM 40239 (ATCC 11989; (Soliveri et al. 1993)) was obtained from the DSMZ culture collection. For analysis of cell-wall composition, a thin-layer-chromatographic procedure was used as described previously (Staneck and Roberts 1974) to determine the isomers of diaminopimelic acid in biomass of K. setae KM-6054T and Kitasatospora MBT63 and MBT66 scraped from solid SFM agar plates grown for a few days at 30 °C.
DNA manipulation and PCR amplification
General DNA manipulations were performed as described previously (Ausubel et al. 1997). PCRs were carried out with Phusion enzyme (Finnzymes, Bioké, Leiden, the Netherlands) as previously described (Colson et al. 2007). Primers (Table S3) were synthesised by Eurogentec (Maastricht, The Netherlands). For PCR of the regions around ssgRA in Kitasatospora, primer pairs MBT63_flank_F/R and MBT66_flank_F/R were used to amplify the region corresponding to nt positions 4438259–4440381 of the K. setae genome, and primer pair Kita_ssgA_F/R was used to amplify the ssgA gene.
Genome sequencing, assembly, annotation, scaffolding and comparisons
DNA isolation was performed as follows. Strains were grown in 50 ml TSBS-YEME (v:v 1:1) with 5 mM MgCl2 and 0.5 % glycine at 30 °C for 48–72 h depending on growth rate. Cells were resuspended in 10 mM NaCl, 20 mM Tris–HCl (pH 8.0), 1 mM EDTA and incubated with lysozyme at 37 °C for 1–30 min until cells were lysed. SDS (0.5 % final concentration) and proteinase K (40 µg) were added and cell extracts incubated at 50 °C for 6 h or overnight. Classical phenol/chloroform extraction was subsequently performed on cell lysates. Extracts were adjusted to 0.3 M NaOAC (pH 5.5) and DNA spooled out with glass rods upon addition of 2 volumes of 96 % ethanol. After washing and drying, DNA preparations were dissolved in TE buffer; DNA quality was verified by SalI digestion and agarose gel electrophoresis.
Illumina/Solexa sequencing on Genome Analyzer IIx and sequencing on PacBio RS were outsourced (BaseClear, Leiden, The Netherlands for Kitasatospora strain MBT66 and Service XS, Leiden, The Netherlands for Kitasatospora MBT63). 100-nt paired-end reads were obtained and the quality of the short reads verified using FastQC (http://www.bioinformatics.bbsrc.ac.uk/projects/fastqc/). Depending on quality, reads were trimmed to various lengths at both ends. Processed raw reads were subsequently used as input for the Velvet assembly algorithm (Zerbino and Birney 2008).
Genomes were annotated using the RAST server (Aziz et al. 2008) with default options. Contigs were also annotated using GeneMark.hmm (Lukashin and Borodovsky 1998) for ORF prediction, BLASTP (Altschul et al. 1990) for putative function prediction and HMMER (Finn et al. 2011) for protein-domain prediction, manually inspected for some (particularly for lantibiotic precursor [lanA] prediction) and visualized using Artemis (Berriman and Rutherford 2003).
The MBT66 genome was scaffolded against reference genomes (K. setae KM-6054T and S. coelicolor A3(2)) using R2CAT (Husemann and Stoye 2010). For genome comparisons using MAUVE (Darling et al. 2010), the MBT66 genome was first reordered as follows: contigs that could be matched against the relevant reference genome using R2Cat were thereby scaffolded into a new Fasta file, and unmatched contigs were added at the end of the Fasta file. Such a re-organized Fasta file was used as input in MAUVE against a reference genome.
Phylogenetic analyses
RecA, RpoB, SsgA and SsgA-like proteins, SapF and 16S rRNA sequences were retrieved from GenBank. Most sequences were published previously (Girard et al. 2013) or provided in the online supplemental information. Sequences were aligned with Mafft (Katoh et al. 2009). Alignments were trimmed for gaps where more than 5 % of the sequences were missing, using the Extractalign tool of the eBioX package (http://www.ebioinformatics.org/ebiox/). Phylogenetic trees were generated using maximum-likelihood algorithms with default parameters as implemented in MEGA version 5 (Tamura et al. 2011). The tree reliability was estimated by bootstrapping with 1,000 replicates (Busarakam et al. 2014). Since groupings supported by poor bootstrap values are not reliable, internal branches with a bootstrap value of less than 50 % were collapsed so as to emphasize the reliable branching patterns.
Data accessibility
The Whole Genome Shotgun projects of Kitasatospora strains MBT63 and MBT66 have been deposited at DDBJ/EMBL/GenBank under the accession numbers JAIZ00000000 and JAIY00000000, respectively. All other accession numbers of the DNA and protein sequences have been made available in the online Supplemental Material.
Abbreviations
- aa:
-
Amino acid
- A2pm:
-
Diaminopimelic acid
- nt:
-
Nucleotide
- SALP:
-
SsgA-like protein
References
Ainsa JA, Bird N, Ryding NJ, Findlay KC, Chater KF (2010) The complex whiJ locus mediates environmentally sensitive repression of development of Streptomyces coelicolor A3(2). Antonie Van Leeuwenhoek 98:225–236
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ (1990) Basic local alignment search tool. J Mol Biol 215:403–410
Angell S, Lewis CG, Buttner MJ, Bibb MJ (1994) Glucose repression in Streptomyces coelicolor A3(2): a likely regulatory role for glucose kinase. Mol Gen Genet 244:135–143
Ausubel FM, Brent R, Kingston RE, Moore DD, Seidman JG, Smith JA, Struhl K (1997) Currents protocols in molecular biology. Wiley, New York
Aziz RK, Bartels D, Best AA, DeJongh M, Disz T, Edwards RA, Formsma K, Gerdes S, Glass EM, Kubal M, Meyer F, Olsen GJ, Olson R, Osterman AL, Overbeek RA, McNeil LK, Paarmann D, Paczian T, Parrello B, Pusch GD, Reich C, Stevens R, Vassieva O, Vonstein V, Wilke A, Zagnitko O (2008) The RAST server: rapid annotations using subsystems technology. BMC Genom 9:75
Bagchi S, Tomenius H, Belova LM, Ausmees N (2008) Intermediate filament-like proteins in bacteria and a cytoskeletal function in Streptomyces. Mol Microbiol 70:1037–1050
Baltz RH (2008) Renaissance in antibacterial discovery from actinomycetes. Curr Opin Pharmacol 8:557–563
Bentley SD, Chater KF, Cerdeno-Tarraga AM, Challis GL, Thomson NR, James KD, Harris DE, Quail MA, Kieser H, Harper D, Bateman A, Brown S, Chandra G, Chen CW, Collins M, Cronin A, Fraser A, Goble A, Hidalgo J, Hornsby T, Howarth S, Huang CH, Kieser T, Larke L, Murphy L, Oliver K, O’Neil S, Rabbinowitsch E, Rajandream MA, Rutherford K, Rutter S, Seeger K, Saunders D, Sharp S, Squares R, Squares S, Taylor K, Warren T, Wietzorrek A, Woodward J, Barrell BG, Parkhill J, Hopwood DA (2002) Complete genome sequence of the model actinomycete Streptomyces coelicolor A3(2). Nature 417:141–147
Berriman M, Rutherford K (2003) Viewing and annotating sequence data with Artemis. Brief Bioinform 4:124–132
Busarakam K, Bull AT, Girard G, Labeda DP, van Wezel GP, Goodfellow M (2014) Streptomyces leeuwenhoekii sp nov., the producer of chaxalactins and chaxamycins, forms a distinct branch in Streptomyces gene trees. Antonie van Leeuwenhoek 105(5):849–861
Celler K, Koning RI, Koster AJ, van Wezel GP (2013) Multidimensional view of the bacterial cytoskeleton. J Bacteriol 195:1627–1636
Chater KF (1972) A morphological and genetic mapping study of white colony mutants of Streptomyces coelicolor. J Gen Microbiol 72:9–28
Chater KF, Losick R (1997) Mycelial life style of Streptomyces coelicolor A3(2) and its relatives. In: Shapiro JA, Dworkin M (eds) Bacteria as multicellular organisms. Oxford University Press, New York, pp 149–182
Claessen D, Rozen DE, Kuipers OP, Sogaard-Andersen L, van Wezel GP (2014) Bacterial solutions to multicellularity: a tale of biofilms, filaments and fruiting bodies. Nat Rev Microbiol 12:115–124
Colson S, Stephan J, Hertrich T, Saito A, van Wezel GP, Titgemeyer F, Rigali S (2007) Conserved cis-acting elements upstream of genes composing the chitinolytic system of streptomycetes are DasR-responsive elements. J Mol Microbiol Biotechnol 12:60–66
Colson S, van Wezel GP, Craig M, Noens EE, Nothaft H, Mommaas AM, Titgemeyer F, Joris B, Rigali S (2008) The chitobiose-binding protein, DasA, acts as a link between chitin utilization and morphogenesis in Streptomyces coelicolor. Microbiology 154:373–382
Craney A, Ozimok C, Pimentel-Elardo SM, Capretta A, Nodwell JR (2012) Chemical perturbation of secondary metabolism demonstrates important links to primary metabolism. Chem Biol 19:1020–1027
Cruz-Morales P, Vijgenboom E, Iruegas-Bocardo F, Girard G, Yanez-Guerra LA, Ramos-Aboites HE, Pernodet JL, Anne J, van Wezel GP, Barona-Gomez F (2013) The genome sequence of Streptomyces lividans 66 reveals a novel tRNA-dependent peptide biosynthetic system within a metal-related genomic island. Genome Biol Evol 5:1165–1175
Darling AE, Mau B, Perna NT (2010) progressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS ONE 5:e11147
Eccleston M, Ali RA, Seyler R, Westpheling J, Nodwell J (2002) Structural and genetic analysis of the BldB protein of Streptomyces coelicolor. J Bacteriol 184:4270–4276
Finn RD, Clements J, Eddy SR (2011) HMMER web server: interactive sequence similarity searching. Nucleic Acids Res 39:W29–W37
Flärdh K, Buttner MJ (2009) Streptomyces morphogenetics: dissecting differentiation in a filamentous bacterium. Nat Rev Microbiol 7:36–49
Foulston LC, Bibb MJ (2010) Microbisporicin gene cluster reveals unusual features of lantibiotic biosynthesis in actinomycetes. Proc Natl Acad Sci U S A 107:13461–13466
Girard G, Traag BA, Sangal V, Mascini N, Hoskisson PA, Goodfellow M, van Wezel GP (2013) A novel taxonomic marker that discriminates between morphologically complex actinomycetes. Open Biol 3:130073
Goodfellow M (2012) Phylum XXVI. Actinobacteria phyl. nov. In: Goodfellow M, Kämpfer P, Busse H-J, Trujillo ME, Suzuki K-I, Ludwig W, Whitman WB (eds) Bergey’s manual of systematic bacteriology, vol 5. The Actinobacteria, parts A and B, 2nd edn. Springer, New York, pp 1–2083
Gourevitch A, Lein J (1955) Production of tetracycline and substituted tetracyclines. US Patent 2,712,517
Heichlinger A, Ammelburg M, Kleinschnitz EM, Latus A, Maldener I, Flärdh K, Wohlleben W, Muth G (2011) The MreB-like protein Mbl of Streptomyces coelicolor A3(2) depends on MreB for proper localization and contributes to spore wall synthesis. J Bacteriol 193:1533–1542
Holmes NA, Walshaw J, Leggett RM, Thibessard A, Dalton KA, Gillespie MD, Hemmings AM, Gust B, Kelemen GH (2013) Coiled-coil protein Scy is a key component of a multiprotein assembly controlling polarized growth in Streptomyces. Proc Natl Acad Sci USA 110:E397–E406
Hopwood DA (2007) Streptomyces in nature and medicine: the antibiotic makers. Oxford University Press, New York
Husemann P, Stoye J (2010) r2cat: synteny plots and comparative assembly. Bioinformatics 26:570–571
Ichikawa N, Oguchi A, Ikeda H, Ishikawa J, Kitani S, Watanabe Y, Nakamura S, Katano Y, Kishi E, Sasagawa M, Ankai A, Fukui S, Hashimoto Y, Kamata S, Otoguro M, Tanikawa S, Nihira T, Horinouchi S, Ohnishi Y, Hayakawa M, Kuzuyama T, Arisawa A, Nomoto F, Miura H, Takahashi Y, Fujita N (2010) Genome sequence of Kitasatospora setae NBRC 14216T: an evolutionary snapshot of the family Streptomycetaceae. DNA Res 17:393–406
Jakimowicz D, van Wezel GP (2012) Cell division and DNA segregation in Streptomyces: how to build a septum in the middle of nowhere? Mol Microbiol 85:393–404
Kämpfer P (2012) Family 1. Streptomycetaceae Waksman and Henrici 1943, 339AL emend. Rainey, Ward-Rainey and Stackebrandt, 1997, 486 emend. Kim, Lonsdale, Seong and Goodfellow 2003b, 113 emend. Zhi, Li and Stackebrandt 2009, 600. In: Goodfellow M, Kämpfer P, Busse H-J, Trujillo ME, Suzuki K-I, Ludwig W, Whitman WB (eds) Bergey’s manual of systematic bacteriology, vol 5. 2nd edn. Springer, New York
Katoh K, Asimenos G, Toh H (2009) Multiple alignment of DNA sequences with MAFFT. Methods Mol Biol 537:39–64
Kawamoto S, Watanabe H, Hesketh A, Ensign JC, Ochi K (1997) Expression analysis of the ssgA gene product, associated with sporulation and cell division in Streptomyces griseus. Microbiology 143:1077–1086
Keijser BJ, Noens EE, Kraal B, Koerten HK, van Wezel GP (2003) The Streptomyces coelicolor ssgB gene is required for early stages of sporulation. FEMS Microbiol Lett 225:59–67
Kirby R (2011) Chromosome diversity and similarity within the Actinomycetales. FEMS Microbiol Lett 319:1–10
Kirby R, Gan TK, Hunter I, Herron P, Tilley E (2008) The genome of Streptomyces rimosus subsp. rimosus shows a novel structure compared to other Streptomyces using DNA/DNA microarray analysis. Antonie Van Leeuwenhoek 94:173–186
Kirby R, Herron P, Hoskisson P (2011) Analysis of developmental gene conservation in the Actinomycetales using DNA/DNA microarray comparisons. Antonie Van Leeuwenhoek 99:159–177
Kodani S, Hudson ME, Durrant MC, Buttner MJ, Nodwell JR, Willey JM (2004) The SapB morphogen is a lantibiotic-like peptide derived from the product of the developmental gene ramS in Streptomyces coelicolor. Proc Natl Acad Sci USA 101:11448–11453
Labeda DP, Goodfellow M, Brown R, Ward AC, Lanoot B, Vanncanneyt M, Swings J, Kim SB, Liu Z, Chun J, Tamura T, Oguchi A, Kikuchi T, Kikuchi H, Nishii T, Tsuji K, Yamaguchi Y, Tase A, Takahashi M, Sakane T, Suzuki KI, Hatano K (2012) Phylogenetic study of the species within the family Streptomycetaceae. Antonie Van Leeuwenhoek 101:73–104
Lewis RA, Laing E, Allenby N, Bucca G, Brenner V, Harrison M, Kierzek AM, Smith CP (2010) Metabolic and evolutionary insights into the closely-related species Streptomyces coelicolor and Streptomyces lividans deduced from high-resolution comparative genomic hybridization. BMC Genom 11:682
Lin YS, Kieser HM, Hopwood DA, Chen CW (1993) The chromosomal DNA of Streptomyces lividans 66 is linear. Mol Microbiol 10:923–933
Ludwig W, Euzeby J, Schumann P, Busse H-J, Trujillo ME, Kämpfer P, Whitman WB (2012) Road map of the phylum Actinobacteria. In: Goodfellow M, Kämpfer P, Busse H-J, Trujillo ME, Suzuki K-I, Ludwig W, Whitman WB (eds) Bergey’s manual of systematic bacteriology, vol 5. 2nd edn. Springer, New York, pp 1–28
Lukashin AV, Borodovsky M (1998) GeneMark.hmm: new solutions for gene finding. Nucleic Acids Res 26:1107–1115
McCormick JR (2009) Cell division is dispensable but not irrelevant in Streptomyces. Curr Opin Biotechnol 12:689–698
Medema MH, Blin K, Cimermancic P, de Jager V, Zakrzewski P, Fischbach MA, Weber T, Takano E, Breitling R (2011) antiSMASH: rapid identification, annotation and analysis of secondary metabolite biosynthesis gene clusters in bacterial and fungal genome sequences. Nucleic Acids Res 39:W339–W346
Merrick MJ (1976) A morphological and genetic mapping study of bald colony mutants of Streptomyces coelicolor. J Gen Microbiol 96:299–315
Noens EE, Mersinias V, Willemse J, Traag BA, Laing E, Chater KF, Smith CP, Koerten HK, van Wezel GP (2007) Loss of the controlled localization of growth stage-specific cell-wall synthesis pleiotropically affects developmental gene expression in an ssgA mutant of Streptomyces coelicolor. Mol Microbiol 64:1244–1259
Omura S, Takahashi Y, Iwai Y, Tanaka H (1982) Kitasatosporia, a new genus of the order Actinomycetales. J Antibiot (Tokyo) 35:1013–1019
Pope MK, Green BD, Westpheling J (1996) The bld mutants of Streptomyces coelicolor are defective in the regulation of carbon utilization, morphogenesis and cell–cell signalling. Mol Microbiol 19:747–756
Pope MK, Green B, Westpheling J (1998) The bldB gene encodes a small protein required for morphogenesis, antibiotic production, and catabolite control in Streptomyces coelicolor. J Bacteriol 180:1556–1562
Shendure J, Lieberman Aiden E (2012) The expanding scope of DNA sequencing. Nat Biotechnol 30:1084–1094
Soliveri J, Vijgenboom E, Granozzi C, Plaskitt KA, Chater KF (1993) Functional and evolutionary implications of a survey of various actinomycetes for homologues of two Streptomyces coelicolor sporulation genes. J Gen Microbiol 139:2569–2578
Soliveri JA, Gomez J, Bishai WR, Chater KF (2000) Multiple paralogous genes related to the Streptomyces coelicolor developmental regulatory gene whiB are present in Streptomyces and other actinomycetes. Microbiology 146:333–343
Soufo HJ, Graumann PL (2003) Actin-like proteins MreB and Mbl from Bacillus subtilis are required for bipolar positioning of replication origins. Curr Biol 13:1916–1920
Staneck JL, Roberts GD (1974) Simplified approach to identification of aerobic actinomycetes by thin-layer chromatography. Appl Microbiol 28:226–231
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S (2011) MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28:2731–2739
Traag BA, van Wezel GP (2008) The SsgA-like proteins in actinomycetes: small proteins up to a big task. Antonie Van Leeuwenhoek 94:85–97
Traag BA, Kelemen GH, Van Wezel GP (2004) Transcription of the sporulation gene ssgA is activated by the IclR-type regulator SsgR in a whi-independent manner in Streptomyces coelicolor A3(2). Mol Microbiol 53:985–1000
van Wezel GP, McDowall KJ (2011) The regulation of the secondary metabolism of Streptomyces: new links and experimental advances. Nat Prod Rep 28:1311–1333
van Wezel GP, van der Meulen J, Kawamoto S, Luiten RG, Koerten HK, Kraal B (2000) ssgA is essential for sporulation of Streptomyces coelicolor A3(2) and affects hyphal development by stimulating septum formation. J Bacteriol 182:5653–5662
van Wezel GP, Krabben P, Traag BA, Keijser BJ, Kerste R, Vijgenboom E, Heijnen JJ, Kraal B (2006) Unlocking Streptomyces spp. for use as sustainable industrial production platforms by morphological engineering. Appl Environ Microbiol 72:5283–5288
Wellington EM, Stackebrandt E, Sanders D, Wolstrup J, Jorgensen NO (1992) Taxonomic status of Kitasatosporia, and proposed unification with Streptomyces on the basis of phenotypic and 16S rRNA analysis and emendation of Streptomyces Waksman and Henrici 1943, 339AL. Int J Syst Bacteriol 42:156–160
Willemse J, Borst JW, de Waal E, Bisseling T, van Wezel GP (2011) Positive control of cell division: FtsZ is recruited by SsgB during sporulation of Streptomyces. Genes Dev 25:89–99
Willey JM, van der Donk WA (2007) Lantibiotics: peptides of diverse structure and function. Annu Rev Microbiol 61:477–501
Willey J, Santamaria R, Guijarro J, Geistlich M, Losick R (1991) Extracellular complementation of a developmental mutation implicates a small sporulation protein in aerial mycelium formation by S. coelicolor. Cell 65:641–650
Xu Q, Traag BA, Willemse J, McMullan D, Miller MD, Elsliger MA, Abdubek P, Astakhova T, Axelrod HL, Bakolitsa C, Carlton D, Chen C, Chiu HJ, Chruszcz M, Clayton T, Das D, Deller MC, Duan L, Ellrott K, Ernst D, Farr CL, Feuerhelm J, Grant JC, Grzechnik A, Grzechnik SK, Han GW, Jaroszewski L, Jin KK, Klock HE, Knuth MW, Kozbial P, Krishna SS, Kumar A, Marciano D, Minor W, Mommaas AM, Morse AT, Nigoghossian E, Nopakun A, Okach L, Oommachen S, Paulsen J, Puckett C, Reyes R, Rife CL, Sefcovic N, Tien HJ, Trame CB, van den Bedem H, Wang S, Weekes D, Hodgson KO, Wooley J, Deacon AM, Godzik A, Lesley SA, Wilson I, van Wezel GP (2009) Structural and functional characterizations of SsgB, a conserved activator of developmental cell division in morphologically complex actinomycetes. J Biol Chem 284:25268–25279
Yang CC, Huang CH, Li CY, Tsay YG, Lee SC, Chen CW (2002) The terminal proteins of linear Streptomyces chromosomes and plasmids: a novel class of replication priming proteins. Mol Microbiol 43:297–305
Zerbino DR, Birney E (2008) Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res 18:821–829
Zhang Z, Wang Y, Ruan J (1997) A proposal to revive the genus Kitasatospora (Omura, Takahashi, Iwai, and Tanaka 1982). Int J Syst Bacteriol 47:1048–1054
Zhu H, Sandiford SK, van Wezel GP (2014a) Triggers and cues that activate antibiotic production by actinomycetes. J Ind Microbiol Biotechnol 41:371–386
Zhu H, Swierstra J, Wu C, Girard G, Choi YH, van Wamel W, Sandiford S, van Wezel GP (2014b) Eliciting antibiotics active against the ESKAPE pathogens in a collection of actinomycetes isolated from mountain soils. Microbiology In the press
Acknowledgments
We are grateful to Eric Miller and Daniel Melton for technical assistance, KB was supported by a scholarship from the Thai Royal, Government, GG by grant 10479 from the Dutch Technology Foundation (STW) to GPvW, and MG by an Emeritus Fellowship from the Leverhulme Trust.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Girard, G., Willemse, J., Zhu, H. et al. Analysis of novel kitasatosporae reveals significant evolutionary changes in conserved developmental genes between Kitasatospora and Streptomyces . Antonie van Leeuwenhoek 106, 365–380 (2014). https://doi.org/10.1007/s10482-014-0209-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10482-014-0209-1