
Abstract
Vascular endothelial growth factor-A (VEGF-A/VEGF) interaction with VEGF receptor 2 (VEGFR2) is key for sprouting angiogenesis in health and disease. VEGF/VEGFR2 signaling promotes endothelial proliferation and migration, as well as the hierarchical organization into leader (tip) and follower (stalk) cells via a dynamic interplay with Notch. Recent studies reveal novel molecular mechanisms to fine-tune VEGF/Notch signaling and tip/stalk cell function during sprouting angiogenesis.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
VEGF/VEGFR2 interaction promotes endothelial cell proliferation, migration, and survival, largely via activation of the phosphoinositide 3-kinase (PI3-K)/Akt and extracellular signal-regulated kinase (ERK) pathways [1]. In addition, VEGF-induced kinase signaling promotes the development of tip cells, which have a specific transcriptional signature and express a variety of proteins that mark their identity [2, 3]. Some of these proteins, such as Delta-like 4 (Dll4), suppress the development of tip cell properties in adjacent stalk cells, by trans-activation of the Notch pathway [4]. Although tip cells encounter highest levels of VEGF, they are generally less proliferative than stalk cells and mainly stimulate sprout migration. Conversely, stalk cells are actively proliferating despite high Notch signaling, which is thought to inhibit proliferation. Thus, anti- and pro-proliferative cues resulting from VEGF/Notch signaling need to be continuously fine-tuned in a spatiotemporal manner, to achieve appropriate cellular responses in angiogenic sprouts. Recent data illuminate novel mechanisms by which such spatiotemporal control is exerted by kinases.
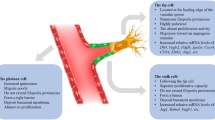
ERK activation occurs via the canonical Ras/Raf pathway but is further enhanced by protein kinase C (PKC), in a manner dependent on diacylglycerol and inositoltrisphosphate-triggered release of Ca2+ from the endoplasmic reticulum (ER) (Fig. 1). In a recent study, it was found that the ER-resident protein transmembrane protein 33 (TMEM33) is required for VEGF-induced Ca2+ release from the ER, and that cytosolic Ca2+ oscillations in tip cells under the control of TMEM33 promote filopodia formation and migration of tip cells [5]. Furthermore, Ca2+ influx potentiates ERK activation and downstream Dll4/Notch signaling, and the knockdown of Tmem33 in zebrafish impaired VEGF-dependent ERK phosphorylation, downstream Notch signaling, and vascular morphogenesis during embryonic development [5]. In another recent report, zebrafish endothelial cells were examined for genes that are transcriptionally activated by VEGF and repressed by Notch, to identify positive-feedback regulators of VEGF/VEGFR2 signaling. A transcript was identified encoding the atypical tetraspanin Tm4sf18 (TM4SF1 in humans). Subsequent studies using zebrafish mutants and knockdown experiments in human endothelial cells revealed that Tm4sf18/TM4SF1 amplifies VEGF/VEGFR2 signaling to ERK and stimulates the rapid selection of tip cells [6]. In the absence of this amplification loop, lower numbers of branching cells and reduced cellularity of newly formed vessels were observed [6]. Since tetraspanins associate with cell-surface proteins including integrins and receptor tyrosine kinases, it is possible that Tm4sf18/TM4SF1 modulates VEGFR2 signaling output directly from the plasma membrane.

Regulatory mechanisms controlling tip/stalk cell biology. VEGF/VEGFR2 signaling activates the PI3-K/Akt and ERK pathways important for proliferation, migration, and the expression of tip cell genes including DLL4. ERK activation is enhanced by PKC, which requires TMEM33-dependent release of Ca2+ from the ER and is further promoted by the positive-feedback regulator TM4SF1. However, excessive ERK activation in tip cells induces expression of the cell cycle inhibitor P21, leading to cell cycle arrest. P21 expression is regulated by FOXO1, a transcription factor that stimulates tip cell polarity and migration while preventing proliferation. While hypoxia promotes the nuclear retention and DNA-binding activity of FOXO1 via Mst1, these events are opposed by PI3-K/Akt and aPKCλ in response to VEGF
The PI3-K/Akt pathway leads to phosphorylation of Forkhead box protein O1 (FOXO1), a member of the FOXO family of transcription factors that are highly responsive to cellular stress and locate in the nucleus when environmental growth factors are low. In such conditions, FOXO1 maintains endothelial quiescence by reducing glycolysis and mitochondrial respiration and by blocking cell proliferation. The cell cycle arrest imposed by FOXO1 is due to expression of cell cycle inhibitors including p21 and p27, as well as the simultaneous suppression of cyclins and c-Myc. FOXO1 phosphorylation in response to growth factor-induced PI3-K/Akt signaling triggers its nuclear export, thus inhibiting FOXO1-dependent gene regulation and alleviating the cell cycle block (Fig. 1). A recent study now shows that the effects of the PI3-K/Akt pathway on FOXO1 are opposed by Mammalian sterile 20-like kinase 1 (Mst1), which phosphorylates FOXO1 at a site different from that used by Akt. Mst1 is activated by cellular stress such as growth factor/nutrient deprivation or hypoxia, which leads to accumulation of reactive oxygen species (ROS) produced by mitochondria (Fig. 1). Mst1-dependent phosphorylation enhances the nuclear import of FOXO1, and augments the transcription of tip cell-, polarity-, and migration‐associated genes, which is induced by FOXO1 in response to hypoxia [7, 8]. In this way, the Mst1-FOXO1 cascade promotes polarized, directional migration of tip cells towards hypoxic regions, and endothelial‐specific deletion of either Mst1 or FOXO1 in mice inhibits tip cell polarization and sprouting angiogenesis [7, 8]. In addition to this mechanism, another recent study unveiled that atypical PKCλ (aPKCλ) also phosphorylates FOXO1, at a site close to but distinct from that recognized by Mst1. Intriguingly, this phosphorylation does not affect the nuclear localization of FOXO1 but impairs its binding to DNA and therefore stimulates c-Myc expression and proliferation [9]. Consequently, endothelial deletion of aPKCλ in mice impairs c-Myc expression and proliferation, both during developmental angiogenesis and in malignant endothelial neoplasms [9]. Thus, multiple kinases jointly regulate FOXO1 function at several levels and therefore shape the balance between cell cycle arrest, migration, and proliferation in response to various environmental stimuli.
Intriguingly, it was recently found that endothelial cell cycle exit imposed by high p21 levels can also result from excessive VEGF stimulation and ERK signaling. Tip cells in sprouting retinal vessels encounter highest concentrations of VEGF and as a result have high levels of ERK signaling, but withdraw from the cell cycle due to the induction of p21 [10]. The cell cycle arrest enhances endothelial sprouting and migration, but eventually blocks angiogenesis because of insufficient proliferation. The cause for this unexpected finding is a bell-shaped dose/response curve to VEGF-induced ERK signaling; either very low or very high levels of active ERK trigger p21-dependent cell cycle arrest. This mechanism explains why tip cells are generally less proliferative despite being exposed to high levels of VEGF, while stalk cells are actively cycling at lower VEGF concentrations and lower ERK activation levels [10].
Together, these studies emphasize the context dependency of VEGF/Notch signaling, the importance of local VEGF fluctuations, and the intricate way in which endothelial cells spatiotemporally control pro- and anti-angiogenic functions of the same stimuli. It will be very important to take these mechanisms into account when considering future VEGF/Notch-targeted therapies for the inhibition or stimulation of angiogenesis in disease settings.
Abbreviations
- aPKC:
-
Atypical PKC
- DAG:
-
Diacylglycerol
- Dll4:
-
Delta-like ligand 4
- ER:
-
Endoplasmic reticulum
- ERK:
-
Extracellular signal-regulated kinase
- FOXO1:
-
Forkhead box protein O1
- IP3:
-
Inositoltrisphosphate
- Mst1:
-
Mammalian sterile 20-like kinase 1
- ROS:
-
Reactive oxygen species
- PI3-K:
-
Phosphoinositide 3-kinase
- PKC:
-
Protein kinase C
- TMEM33:
-
Transmembrane protein 33
- VEGF:
-
Vascular endothelial growth factor
- VEGFR2:
-
VEGF receptor 2
References
Simons M, Gordon E, Claesson-Welsh L (2016) Mechanisms and regulation of endothelial VEGF receptor signalling. Nat Rev Mol Cell Biol 17(10):611–625
del Toro R, Prahst C, Mathivet T, Siegfried G, Kaminker JS, Larrivee B, Breant C, Duarte A, Takakura N, Fukamizu A, Penninger J, Eichmann A (2010) Identification and functional analysis of endothelial tip cell-enriched genes. Blood 116(19):4025–4033
Dallinga MG, Yetkin-Arik B, Kayser RP, Vogels IMC, Nowak-Sliwinska P, Griffioen AW, van Noorden CJF, Klaassen I, Schlingemann RO (2018) IGF2 and IGF1R identified as novel tip cell genes in primary microvascular endothelial cell monolayers. Angiogenesis 21(4):823–836
Jakobsson L, Bentley K, Gerhardt H (2009) VEGFRs and Notch: a dynamic collaboration in vascular patterning. Biochem Soc Trans. 37(Pt 6):1233–1236
Savage AM, Kurusamy S, Chen Y, Jiang Z, Chhabria K, MacDonald RB, Kim HR, Wilson HL, van Eeden FJM, Armesilla AL, Chico TJA, Wilkinson RN (2019) tmem33 is essential for VEGF-mediated endothelial calcium oscillations and angiogenesis. Nat Commun 10(1):732
Page DJ, Thuret R, Venkatraman L, Takahashi T, Bentley K, Herbert SP (2019) Positive feedback defines the timing, magnitude, and robustness of angiogenesis. Cell Rep 27(11):3139–3151
Kim YH, Choi J, Yang MJ, Hong SP, Lee CK, Kubota Y, Lim DS, Koh GY (2019) A MST1-FOXO1 cascade establishes endothelial tip cell polarity and facilitates sprouting angiogenesis. Nat Commun 10(1):838
Fukumoto M, Kondo K, Uni K, Ishiguro T, Hayashi M, Ueda S, Mori I, Niimi K, Tashiro F, Miyazaki S, Miyazaki JI, Inagaki S, Furuyama T (2018) Tip-cell behavior is regulated by transcription factor FoxO1 under hypoxic conditions in developing mouse retinas. Angiogenesis 21(2):203–214
Riddell M, Nakayama A, Hikita T, Mirzapourshafiyi F, Kawamura T, Pasha A, Li M, Masuzawa M, Looso M, Steinbacher T, Ebnet K, Potente M, Hirose T, Ohno S, Fleming I, Gattenlöhner S, Aung PP, Phung T, Yamasaki O, Yanagi T, Umemura H, Nakayama M (2018) aPKC controls endothelial growth by modulating c-Myc via FoxO1 DNA-binding ability. Nat Commun 9(1):5357
Pontes-Quero S, Fernández-Chacón M, Luo W, Lunella FF, Casquero-Garcia V, Garcia-Gonzalez I, Hermoso A, Rocha SF, Bansal M, Benedito R (2019) High mitogenic stimulation arrests angiogenesis. Nat Commun 10(1):2016
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Margadant, C. Positive and negative feedback mechanisms controlling tip/stalk cell identity during sprouting angiogenesis. Angiogenesis 23, 75–77 (2020). https://doi.org/10.1007/s10456-020-09706-0
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10456-020-09706-0