
Abstract
Ischemic diseases such as stroke and proliferative retinopathy are characterized by hypoxia-driven release of angiogenic factors such as vascular endothelial growth factor (VEGF). However, revascularization of the ischemic areas is inadequate, resulting in impaired neuro-vascular function. We aim to examine the vascular protective effects of candesartan, an angiotensin receptor blocker, in an ischemic retinopathy mouse model. Vascular density, number of tip cells, and perfusions of capillaries were assessed. Activation of Muller glial cells and levels of peroxynitrite, VEGF, VEGFR2, inducible nitric oxide synthase, hemeoxygenase-1 (HO-1) were assessed. Proangiogenic effects of candesartan were examined in human endothelial cells (EC) that were cultured in normoxia or hypoxia and transduced with siRNA against HO-1. Candesartan (1 mg/kg) and (10 mg/kg) decreased hypoxia-induced neovascularization by 67 and 70 %, respectively. Candesartan (10 mg/kg) significantly stimulated the number of tip cells and physiological revascularization of the central retina (45 %) compared with untreated pups. The effects of candesartan coincided with reduction of hypoxia-induced Muller glial activation, iNOS expression and restoration of HO-1 expression with no significant change in VEGF levels. In vitro, silencing HO-1 expression blunted the ability of candesartan to induce VEGF expression under normoxia and VEGFR2 activation and angiogenic response under both normoxia and hypoxia. These findings suggest that candesartan improved reparative angiogenesis and hence prevented pathological angiogenesis by modulating HO-1 and iNOS levels in ischemic retinopathy. HO-1 is required for VEGFR2 activation and proangiogenic action of candesartan in EC. Candesartan, an FDA-approved drug, could be repurposed as a potential therapeutic agent for the treatment of ischemic diseases.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Ischemic diseases such as stroke and proliferative retinopathy are characterized by hypoxia-driven release of angiogenic factors such as vascular endothelial growth factor (VEGF) [1]. However, inadequate revascularization of the ischemic areas is resulting in impaired neuro-vascular function and overall damage. Identifying new therapeutics to promote reparative angiogenesis will counteract the ischemic insult and prevent further pathological neovascularization observed in proliferative retinopathy [2]. Ischemic retinopathy including retinopathy of prematurity, retinal vein occlusion and diabetic retinopathy is a potentially blinding disorder that affects premature infants and working-age adults [3, 4]. The pathological progression of ischemic retinopathy is characterized by an initiating event of retinal capillary loss, leading to a poorly controlled process of retinal neovascularization toward the vitreous but not in the ischemic areas [4]. The mainstay treatment for retinal neovascularization is limited to laser photocoagulation, a strategy that does not restore revascularization of the retina. Therefore, there is a great need to identify new and effective therapeutics to harness reparative angiogenesis and prevent retinal ischemia.
The renin–angiotensin system (RAS) is classically recognized for its role in the control of systemic blood pressure. However, local production of retinal Angiotensin II suggested a role for Angiotensin II that is independent of the systemic circulation [5]. While Angiotensin II has been shown to promote endothelial cell migration and angiogenesis by inducing angiogenic growth factors [6, 7], blockers of Angiotensin II type 1 AT1 receptors (ARBs) can also modulate angiogenesis [8]. We recently demonstrated that treatment with candesartan induced a pro-angiogenic state, stimulated VEGF/VEGFR2 and vascular protection in an ischemic stroke model [9–11]. In contrast, previous studies using models of ischemic retinopathy showed that ARBs can prevent pathological angiogenesis by reducing VEGF levels [12, 13] or VEGR2 activation [14]. To clarify ARBs effect on the VEGF/VEGFR2 angiogenic pathway, our study will identify the molecular mechanisms by which candesartan exerts its vascular protective effects in ischemic retinopathy.
ARBs could be a potential strategy to selectively inhibit pathological neovascularization and promote physiological revascularization of the retina [5]. Prior studies have demonstrated the detrimental role of the inducible nitric oxide synthase (iNOS) expression [15] and excessive nitrative stress that induced retinal endothelial cell death and inhibited physiological vascularization of the ischemic areas [16–19]. Avascularization of the ischemic retina can be aggravated also by deficiency of other proangiogenic factors. Hemeoxygenase enzyme (HO-1) has emerged as an important player in wound healing and tumor angiogenesis and the angiogenic response in isolated retinal endothelial cells (EC) [20, 21]. However, little is known on its expression pattern and its potential role in modulating VEGF signal in ischemic retinopathy. Here, we examined the effect of candesartan on nitrative stress and VEGF/VEGFR2 as well as other molecular mediators including expression of iNOS and HO-1 in ischemic retinopathy and cell culture models.
Materials and methods
Animal model
All the animal studies were performed using C57Bl/6J mice from Jackson Laboratories and conducted in accordance with the animal protocols of the Charlie Norwood Veterans’ Affairs Medical Center Animal Care and Use Committee.
Ischemic retinopathy mouse model was induced by exposing neonatal mice along with their dam to 70 % oxygen (hyperoxia) from postnatal day p7 to p12. The pups were brought back to room air (relative hypoxia) and treated with candesartan (a generous gift from AstraZeneca; dissolved in saline, sodium carbonate solution), 1 mg/kg day, i.p., or 10 mg/kg body weight/day, i.p., from p12 to p17 (21 % oxygen). The experimental groups include hypoxia, hypoxia-candesartan, normoxia-candesartan and untreated normoxic pups served as a control group. Pups were deeply anesthetized by IP injection of tribromoethanol (Avertin—240 mg/kg). One eye was enucleated and fixed in 2 % paraformaldehyde overnight to be flat-mounted. For the other eye, retinas were isolated and snap-frozen for biochemical assays.
Analysis of retinal revascularization and pathological neovascularization
Retinal vascular distribution was analyzed using retinal flat mounts labeled with the red fluorescent Alexa Fluor® 594 isolectin GS-IB4 conjugate (Molecular Probes, Life Technology, Grand Island, NY, USA). To examine perfusion of retinal capillaries, fluorescein isothiocyanate-dextran 2 million (FITC-Dextran, 50 mg/ml) [Sigma-Aldrich, MO, USA] was administered at 100 μl i.p. for each pup 30 min before killing. Retinas were viewed and imaged with fluorescence or confocal Axiobserver Zeiss Microscope (Germany). The areas of capillary dropout at p12 and retinal neovascularization were assessed on p17 as described previously [22, 23]. The number of tip cells was counted in isolectin-stained flat-mounted retinas at the border of the central capillary dropout area using a confocal Zeiss microscope. Tip cells were identified by morphology as described previously [24].
Detection of nitrotyrosine
Relative amounts of proteins nitrated on tyrosine were measured using slot–blot techniques as described previously [22]. Briefly, retinal lysates were immobilized onto nitrocellulose membrane by using a slot–blot microfiltration unit (Bio-Rad, Hercules, CA, USA). Nitrotyrosine was detected using a polyclonal anti-nitrotyrosine antibody (Millipore, Temecula, CA, USA) followed by peroxidase-labeled goat anti-mouse IgG and ECL. Relative levels of nitrotyrosine immunoreactivity were determined by densitometry software (Alpha Innotech).
Measurement of retinal nitrite levels
Nitric oxide (NO) production was determined indirectly by measuring the levels of nitrite, the oxidized products of NO, in the supernatant of phosphate-buffered saline (PBS) retinal homogenate by modified Greiss Reagent assay as described before [25]. Briefly, 210 μl of homogenate were incubated with nitrate reductase enzyme (10 mU) and NADPH (12.5 mmol/L) for 30 min at 37 °C. Then the total nitrite in each sample was determined by addition of 200 mU of l-glutamate dehydrogenase, 100 mmol/L NH4Cl, and freshly prepared 4 mmol/L of α-ketoglutarate. The mixture was incubated at 37 °C for 10 min followed by addition of 250 μl of Greiss Reagent and incubation for another 5 min at 37 °C. The absorbance at 543 nm was recorded, and concentrations of nitrite were calculated and expressed as mmol/mg protein.
Determination of retinal lipid peroxides
The assay was performed on retinal lysates as described before [22]. Briefly, retinal lysate is reacted with 20 % acetic acid, 8 % SDS and thiobarbituric acid at 95 °C for 60 min, and the reaction was cooled down on ice. The samples were centrifuged, and the supernatant was extracted with n-butanol and pyridine (15:1, respectively), and the absorbance of the organic solvent layer measured at 532 nm. The Bradford assay (Bio-Rad, Hercules, CA, USA) was performed to determine the protein concentration of the retinal lysate. Lipid peroxide level was expressed in nmole MDA/mg total protein.
Quantitative real-time PCR
The One-Step qRT-PCR kit (Invitrogen) was used to amplify 10 ng retinal mRNA, and quantification was performed as described previously [23]. PCR primers designed to amplify VEGF, TRX and HO-1 were purchased from Integrated DNA Technologies Inc., (IDT, Coralville, IA, USA). Quantitative PCR was performed using a Realplex Master cycler (Eppendorf, Germany) using primers listed in (supplementary table-1). Expression of TRX, VEGF or HO-1 was normalized to the 18S level and expressed as relative expression to control.
Western blot analysis
Retina lysate (30 µg) proteins were boiled in Laemmli buffer at 95 °C for 5 min and separated by electrophoresis and transferred onto nitrocellulose membrane and blocked with 5 % (w/v) nonfat milk for 1 h. The nitrocellulose membranes were then incubated with antibodies including VEGF (Millipore, Billerica, MA, USA, 1:1,000), iNOS, HO-1, pVEGFR2, VEGFR2 (Cell Signaling Technology, Danvers, MA, USA, 1:1,000) in phosphate-buffered saline overnight at 4 °C. After washing, the membranes were incubated with horseradish peroxidase (HRP)-conjugated secondary antibodies and detected with enhanced chemiluminescence (Thermoscientific, Rockford, IL, USA). The films were scanned, and band intensities (optical density) were quantified using imageJ software.
Immunohistochemistry
Frozen retinal cryo-sections (10-µm thickness) were blocked in goat serum and incubated with primary antibodies including iNOS (Cell Signaling, 1:200), CD31 (BD Bioscience Pharmingen, San Diego, CA, USA, 1:100), GFAP (Affinity Bioreagents, Rockford, IL, USA; 1:200) glutamine synthetase (Millipore, Billerica, MA, USA; 1:200), followed by Texas red or Oregon green conjugated goat anti-rabbit or goat anti-mouse (Invitrogen, Carlsbad, CA, USA; 1:500) secondary antibodies. Mounting solution (Vector laboratories, Burlingame, CA, USA) was added, and the slides were sealed using a cover slip. Images were taken by a microscope (AxioObserver.Z1; Zeiss, Germany) under 20× magnification using Axiovision software.
Cell culture
Primary cultures of human retinal EC and supplies were purchased from Cell Systems Corporation (Kirkland, WA, USA). Experiments were performed using cells between passages (4–6). Cells were switched to serum-free medium 6 h prior to stimulation with candesartan. Pilot studies demonstrated that (1 μg/ml) was the sub-maximal dose to induce angiogenic response of candesartan. For treatment, cells were switched to serum-free medium for 4–6 h followed by 24-h candesartan treatment (1 μg/ml). Cells were incubated in a mixture of 5 % CO2, 21 % O2 at 37 °C (normoxia) or balanced N2 in a humidified incubator inside a sealed anaerobic chamber at 1 % O2 (hypoxia), and levels of oxygen were monitored with a sensitive sensor (ProOx model C21; BioSpherix, Lacona, NY, USA). Cells were harvested to detect protein expression using WB or subjected to tube formation assay.
Silencing HO-1 expression
Transfection of human EC was performed using Amaxa nucleofector and a kit for primary EC according to the manufacturer protocol (Lonza, Germany). Optimization experiments that were performed showed that T005-program and (500 ng) of HO-1 siRNA (Santa Cruz) gave the maximum transfection efficacy for EC. Cells suspended in a nucleofector mixture with the siRNA and pmaxGFP were zapped and left 12 h in complete medium to recover before experiments. Transfection efficiency was 60–80 % as indicated by number of GFP expressing cells (data not shown) and Western blots for HO-1 expression.
Tube formation
The alignment of EC into tube-like structures was assessed using Matrigel™ tube formation assay, as described previously [11]. Transfected cells with either scrambled or siRNA were randomized to receive treatment with candesartan (1 μg/ml) or vehicle. The harvested cells were incubated in normoxia or a hypoxia chamber for the designated time and suspended in a mixture of medium-199 (life technologies) and growth factor-reduced Matrigel (BD biosciences) in a ratio of 70:30 (100 µl/well). The mixture was quickly transferred into a 96-well plate and allowed to solidify. Approximately 5 × 104 cells in a volume of 100 µl were seeded into each quadruplicate well. Images were captured 24 h after seeding at an objective lens magnification of 10×. Endothelial cells that had aligned to form >90 % closed structures were considered as tubes and counted in a blinded fashion.
Data analysis
The results were expressed as mean ± SE. Two-way analysis of variance (ANOVA) followed by Bonferroni multiple comparison test was used for testing differences among all the multiple experimental groups and for testing the interaction between the level of oxygen (normoxia vs. hypoxia) across the animal groups (untreated vs. candesartan) and between the presence or absence of HO-1 expression across candesartan-treated or untreated groups (for the in vitro studies). For morphological studies, two-sided Student’s t test was used for testing differences between two experimental groups (hypoxia vs. H+ candesartan). Significance was defined as p < 0.05.
Results
Candesartan prevented retinal neovascularization and improved capillary perfusion in an ischemic retinopathy model
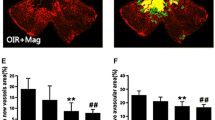
Relative area of tuft, abnormal blood vessels that grow into the vitreous from the retina away from retinal capillary plexuses, is an accepted index for determining the pathological neovascularization. Treatment with candesartan 1 mg/kg (supplementary figure-1, A-B-C) and 10 mg/kg (Fig. 1a, b, f) significantly decreased the pathological tufts by 67 and 70 %, respectively, in the retinal mid-periphery compared with untreated hypoxia. We examined capillary perfusion assessed by injecting FITC-dextran (green) followed by isolectin staining-B4 (red) in flat-mounted retinas. Treatment with candesartan (10 mg/kg) improved capillary perfusion in hypoxic pups compared with untreated mice (Fig. 1c, d). In normoxia, candesartan did not induce alteration of capillary perfusion assessed by FITC-Dextran (supplementary figure-2) or branching density assessed by isolectin-B4 (Fig. 1e).

Candesartan prevented the hypoxia-induced pathological retinal neovascularization. a, b Representative images of flat-mounted retinas of animals subjected to normoxia or hypoxia and treated with candesartan on p17 and labeled with isolectin-B4 (red). c, d Higher magnification confocal images (×63) of retinas perfused with FITC-Dextran (green) and labeled with isolectin-B4 to examine vascular density and the neovascularization. In hypoxia, areas of tufts, a pathological bulb-like structure of new micro-blood vessels grew toward the vitreous far away from the retinal capillary plexus, are higher in the peripheral retinal vasculature with less perfusion at the central areas (c, arrows) compared with those exposed to hypoxia treated with candesartan (10 mg/kg/day, i.p.) (d, arrows). This indicates that candesartan had a reparative response stimulating intra-retinal angiogenesis in the ischemic retinal fields. e Quantification data of vascularized zones normalized to the whole retinal area and vessel density as measured by branch point number in randomized selected field. As the bar chart indicates, the vascular density significantly dropped in hypoxia but not in hypoxia after treatment with candesartan. f Quantification data of tufts (neovascular) area normalized to total the retina area. Tufts area reduced by ~70 % in hypoxia-candesartan compared with hypoxia. p Postnatal, i.p. intraperitoneal, n = 10–12/group, *p < 0.05 versus the rest of groups
Candesartan stimulated tip cells and revascularization of the central ischemic retina
Decrease in capillaries dropout (free) retinal areas during relative hypoxia is an index of physiological retinal revascularization. As shown in (Fig. 2a, b, d), candesartan (10 mg/kg) stimulated reparative angiogenesis evident by 45 % reduction in capillary dropout area compared with untreated hypoxic controls. Moreover, treatment with candesartan stimulated the number of tip cells at the border of capillary dropout toward central retinal, an index of physiological angiogenesis, compared with the untreated hypoxic controls (Fig. 2c, e). Interestingly, treatment with candesartan (1 mg/kg) tended to decrease capillary dropout area (20 %) but did not reach statistical significance (Supplementary figure-1 D). These results suggest that candesartan at (10 mg/kg/day) significantly inhibited the pathological neovascularization and promoted the reparative physiological angiogenesis. Since the goal of the study was to investigate the role of ARBs in physiological angiogenesis to repair the retina, the rest of the studies utilized candesartan 10 mg/kg treatment regimen to explore the possible molecular mechanisms involved.

Candesartan stimulated number of tip cells and enhanced reparative angiogenesis. a, b Representative images of flat-mounted retinas of p17 animals subjected to hypoxia or hypoxia treated with candesartan (10 mg/kg/day, i.p.) and labeled with isolectin-B4 (red). c Higher confocal magnified image (×63) of retinas labeled with isolectin B4 of the part corresponding to b showing tip cells that grow toward the center of the retina to guide newly formed retinal capillaries. d Quantitative data of the capillary dropout area that were normalized to the total retina area showing that candesartan treatment reduced capillary dropout area by 45 % in comparison with that of untreated mice. e Quantitative data of the number of sprouting endothelial cell forming tip cells show that treatment with candesartan (10 mg/kg) significantly increased the number of tip cells that are significantly higher among those treated with candesartan compared with untreated hypoxia. *p < 0.05 versus hypoxia (n = 10–12/group)
Candesartan blocked hypoxia-induced retinal oxidative and nitrative stress
In hypoxia, elevated NO levels become toxic when it interacts with superoxide anion, resulting in the production of the oxidant peroxynitrite. As shown in (Fig. 3a), hypoxia significantly induced nitrotyrosine formation (fourfold) compared with normoxic controls and candesartan-treated normoxic controls. Treatment with candesartan (10 mg/kg) completely blocked hypoxia-induced nitrotyrosine formation. We next examined the formation of NO by determining its stable metabolite, nitrite, using Greiss Reagent. As shown in (Fig. 3b), hypoxia induced a 4.6-fold increase in total nitrite levels compared with normoxic-untreated and candesartan-treated controls. Treatment with candesartan (10 mg/kg) completely blocked hypoxia-induced nitrite formation. We next examined lipid peroxidation as a general measure for oxidative stress. Candesartan (10 mg/kg) inhibited hypoxia-induced increases in lipid peroxidation (Fig. 3c).

Candesartan blocked hypoxia-induced oxidative and nitrative stress via suppression of iNOS. a Representative image of slot–blot and quantification for nitrotyrosine, the footprint of peroxynitrite. Hypoxia significantly induced nitrotyrosine formation compared with the normoxic control group. Candesartan treatment (10 mg/kg/day, i.p.) significantly reduced the nitrotyrosine compared with untreated hypoxia. b Nitrate level detected by Griess reagent, which is an indirect measure of nitric oxide production in the retina, shows a significant increase in nitrate in hypoxia compared with the control groups, which was blocked by candesartan treatment. c Lipid peroxidation detected by TBARs shows a significant increase in oxidative stress in hypoxia compared with the control groups, which was blocked by candesartan treatment. d Representative image and statistical analysis of Western blot for iNOS normalized to β-actin showing that iNOS expression is significantly increased in hypoxia compared with normoxia and normoxia-candesartan groups. Treatment with candesartan (10 mg/kg/day, i.p.) reduced iNOS significantly compared with untreated pups. OD optical density, NY nitrotyrosine. *p < 0.05 versus controls, n = 4–6/group
Candesartan ameliorated hypoxia-induced iNOS expression and glial Muller cell activation
To characterize the sources of nitrative stress at the cell level in the retina and whether candesartan treatment can modify the effect, we examined the expression of iNOS. As shown in (Fig. 3d), hypoxia induced iNOS expression by 1.8-fold compared with untreated normoxic controls and administration of candesartan significantly reduced iNOS expression back to normal. We next examined localization of iNOS and GFAP, a marker for activated glial Müller cells using immunohistochemistry. As shown in (Fig. 4a), hypoxia induced iNOS expression within ganglion cell layer (GCL), inner nuclear layers (INL) and outer plexiform layers (yellow arrows) where retinal capillaries and nuclei of Muller cells reside. Activation of the main glia in the retina, Muller cells, is an index of oxidative and nutritional stress. As shown in (Fig. 4b), hypoxia induced Muller glial cell activation as indicated by radial filamentous expression of GFAP compared with controls. Treatment with candesartan (10 mg/kg) markedly reduced the activation of Muller cells compared with the untreated hypoxia groups. Co-localization studies using iNOS (green) with glutamine synthetase, Muller cells marker (red) showed localization (yellow) within INL and plexiform layer, suggesting that activated Muller cells contribute to nitrative stress in response to hypoxia. Immunostaining of iNOS (green) with CD31 (red), an endothelial cell marker, showed co-localization (yellow) within capillaries in GCL and deep retina capillaries in the inner retina layer (Supplementary figure-3). Together, inhibition of iNOS and nitrative stress may contribute to the mechanism by which candesartan (10 mg/kg) stimulated intraretinal angiogenesis and prevented pathological neovascularization in ischemic retinopathy.

Candesartan ameliorated hypoxia-induced Muller cell activation and suppressed iNOS expression. a Representative images of immunostained retinas with iNOS (green), showing higher expression in hypoxia than normoxia or hypoxia-candesartan. b Representative images of retinas from different treatment groups stained with GFAP (green), a marker for stressed Muller cells. In hypoxia, Muller cell activation is remarkably higher compared with normoxia or normoxia-candesartan and the hypoxic retinas treated with candesartan. GCL Ganglion cell layer, IPL inner plexiform layer, INL inner nuclear layer, ONL outer nuclear layer, DAPI diamidino-2-phenylindole, GFAP glial fibrillary acidic protein. Similar findings were observed in three additional retinas
Candesartan stimulated VEGFR2 activation but did not alter hypoxia-induced VEGF expression
We next examined VEGF expression, at both the mRNA and protein levels at p14. Exposing the pups to relative hypoxia induced retinal VEGF expression by 2.8-fold at mRNA level (Fig. 5a) and 1.9-fold at protein level (Fig. 5b, c) and compared with normoxic controls. Treatment with candesartan did not statistically alter VEGF levels at mRNA or protein levels compared with untreated hypoxia. Under hypoxia, candesartan induced VEGFR2 activation compared with untreated controls (Fig. 5d, e). Under normoxia, candesartan tends to increase VEGF at the mRNA and protein levels; however, it did not reach statistical significance assessed by two-way ANOVA compared with untreated controls. The increases in VEGF in normoxia did not result in alteration of vascular density (Fig. 1e) or retinal capillary perfusion assessed by FITC-dextran perfusion (Supplementary figure-2).

Candesartan stimulated VEGFR2 activation but did not alter VEGF level in hypoxia. a Gene expression of VEGF mRNA normalized to 18 SmRNA of four groups. b, c Representative images of Western blot and statistical analysis for the protein expression of VEGF normalized to β-actin. Candesartan treatment and hypoxia significantly increased the protein expression of VEGF compared with normoxia. Treatment with candesartan during hypoxia did not alter VEGF expression compared with hypoxia. d, e Representative image of Western blot and statistical analysis for VEGFR2 activation showing that hypoxia induced phosphorylation of VEGFR2, an effect that was further maintained by candesartan. Two-way ANOVA showed a significant interaction between candesartan and hypoxia in retinal VEGFR2 activation and VEGF mRNA levels but not at VEGF protein level, *p < 0.05, n = 4–7/group
Candesartan restored retinal hemeoxygenase-1 expression in hypoxia
We next examined the potential impact of candesartan treatment on the expression of antioxidants, thioredoxin (TRX) and hemoxygenase-1 (HO-1). As shown in (Fig. 6a), hypoxia induced retinal total TRX expression 1.45-fold compared with the untreated and treated controls. Treatment with candesartan did not significantly alter TRX level compared with hypoxia alone. While hypoxia did not alter HO-1 expression at the mRNA level (Fig. 6b), it significantly reduced HO-1 at the protein level (Fig. 6c, d) compared with untreated normoxic controls. Treatment with candesartan induced HO-1 mRNA by 1.7-fold in normoxia and 2.4-fold in hypoxia, respectively (Fig. 6b). In parallel, candesartan enhanced HO-1 protein expression by approximately twofold in both normoxia and hypoxia, compared with untreated normoxia (Fig. 6c, d). These results suggest that restoration of HO-1 expression by candesartan may contribute to enhanced angiogenic response apart from hypoxic insult.

Candesartan restored hemeoxygenase-1 expression in hypoxia. Quantitative data of gene expression of the antioxidants. a Total thioredoxin (TRX) mRNA and b hemoxygenase (HO-1) mRNA levels. In hypoxia, TRX mRNA expression significantly increased, but there was no change in HO-1 compared with normoxia. Treatment with candesartan partially but not significantly reduced TRX level compared with hypoxia alone. c, d Representative immunoblots and relative density showing that hypoxia significantly reduced HO-1 expression compared with normoxia. Treatment with candesartan significantly induced HO-1 mRNA and protein levels in normoxia and hypoxia. Two-way ANOVA showed significant interaction between candesartan and hypoxia in retinal HO-1 protein level, *p < 0.05 versus control; # p < 0.05 versus hypoxia, n = 6–8/group
Silencing HO-1 expression induced VEGF expression under hypoxia
In order to examine the role of HO-1 in candesartan-induced proangiogenic response, we used small interfering RNA (siRNA) to target HO-1 in human EC under normoxia and hypoxia. Silencing HO-1 expression was evident by 72 % reduction in siRNA-transduced cells compared with scrambled-transduced cells under normoxia (Fig. 7a, b) and under hypoxia (Fig. 7d, e). Similar to in vivo, candesartan induced expression of HO-1 by 1.8-fold in the scrambled-transduced EC compared with untreated group under normoxia (Fig. 7a, b) as well as hypoxia (Fig. 7d, e). Under normoxia, candesartan tended to increase VEGF expression but did not reach statistical significance, and silencing HO-1 did not alter VEGF expression in scrambled-transduced EC compared with untreated cells (Fig. 7c). Under hypoxia, silencing HO-1 induced VEGF expression by 1.5-fold in the scrambled-EC, and treatment with candesartan did not stimulate VEGF expression compared with hypoxia controls (Fig. 7f). These results recapitulate our observation in vivo, where retinas exposed to hypoxia alone demonstrated less HO-1 and increases in VEGF expression and that candesartan did not stimulate VEGF expression under hypoxia (Figs. 5, 6).

Candesartan enhanced HO-1 expression under normoxia and hypoxia in retinal endothelial cells. Representative images and quantification of HO-1 and VEGF expression levels after 24 h in normoxia (a–c), and hypoxia (d–f) in the presence or absence of candesartan at (1 μg/ml). Candesartan treatment increased levels of HO-1 in normoxia and hypoxia scramble group but not in siRNA group. Candesartan treatment tended to increase VEGF expression in normoxia but not in hypoxia. Scr-Ctrl scramble-control, Scr-Cand scramble-candesartan, siRNA-Ctrl siRNA control, siRNA-Cand siRNA candesartan. Two-way ANOVA showed significant interaction between candesartan and gene silencing in HO-1 protein level in EC, *p < 0.05 versus Scr-Ctrl, n = 3–6/group
Silencing HO-1 expression blunted candesartan-induced VEGFR2 and endothelial cell alignment into tube-like structures
We next examined the impact of silencing HO-1 expression on candesartan-mediated angiogenic response under both normoxia and hypoxia. As shown in (Fig. 8a, b), treatment of the scrambled-transduced EC with candesartan enhanced significantly the phosphorylation of VEGFR2 under both normoxia and hypoxia. Silencing HO-1 expression mitigated candesartan effect under both normoxia and hypoxia. Candesartan treatment also induced the alignment of scrambled-transduced EC into tube-like structures (representative is shown in supplementary figure-4). Candesartan induced tube formation by threefold and fourfold compared with untreated controls under normoxia and hypoxia, respectively (Fig. 8e, f). Silencing HO-1 expression blunted candesartan-induced EC alignment into tube-like structure under both normoxia and hypoxia. These results clearly demonstrate that HO-1 is required for candesartan-mediated VEGFR2 activation and angiogenic response in hypoxic retina and EC.

Candesartan enhanced VEGFR2 activity and stimulated proangiogenic effect. Representative images of Western blot of the pVEGFR2 (on the same membrane in different positions) and their statistical analysis for the protein expression in normoxia (a, b) and hypoxia (c, d), respectively, normalized to the corresponding total-VEGFR2. Treatment with candesartan significantly enhanced the VEGF-R2 activity in normoxia and hypoxia but not with siRNA in both. e, f Quantitative charts of tube-like structures formed in the same treatment groups (corresponding representative images are shown in supplementary Fig. 3). In normoxia, silencing of HO-1 does not modify tube-like structures compared with controls, but candesartan treatment (1 μg/ml) significantly increased (threefold) the effect. b In response to hypoxia, a significant (twofold) increase in tube-like structure compared with normoxia in controls. Silencing of HO-1 significantly (twofold) reduces endothelial cell-tube formation, and candesartan significantly (4.5-fold) abolished the effect compared with controls. Silencing of HO-1 in the presence of candesartan significantly (1.9-fold) reduced tube-like structures compared with controls, indicating that candesartan angiogenic response is HO-1 dependent. Two-way ANOVA showed significant interaction between candesartan and gene silencing in VEGFR2 activation in EC, *p < 0.05 versus other groups, n = 3–6/group
Discussion
Retinal ischemia and the subsequent secretion of angiogenic factors including VEGF are thought to be common precursors to vitreo-retinal neovascularization. Although targeting VEGF and other angiogenic factors has proven effective in preventing pathological vitro-neovascularization, the challenge remains to identify strategies to vascularize the ischemic retina and to maintain the homeostasis of retina vasculature. The findings from this study support the vascular protective effects of candesartan as follows: Candesartan stimulated the revascularization of the central retina and enhanced reparative angiogenesis with subsequent inhibition of hypoxia-induced pathological retinal neovascularization (Figs. 1, 2). The mechanism involves reducing glial activation and nitrative stress via inhibiting iNOS and stimulating HO-1 expression (Figs. 3, 6). HO-1 expression is required for VEGFR2 activation and alignment of EC into tube-like structures under hypoxic condition (Figs. 7, 8). We believe this is the first report to delineate the role of HO-1 in mediating the vascular protective effects of candesartan in ischemic retinopathy.
While several studies indicate that ARBs could inhibit pathological neovascularization and tumor angiogenesis, work with myocardial infarction and cerebral ischemia point to a pro-angiogenic role of ARBs (reviewed in [8]). ARBs have been shown to prevent pathological angiogenesis by decreasing VEGF levels [12, 13] or VEGR2 activation [14]. In agreement, our results showed that lower dose of candesartan (1 mg/kg/day, i.p.) prevented pathological neovascularization; however, it did not stimulate reparative angiogenesis (Supplementary Figure-1). Here using an ischemic retinopathy model, we demonstrate that candesartan at (10 mg/kg/day, i.p.) stimulates reparative angiogenesis as it enhances physiological revascularization of the central retina and hence mitigates retinal ischemia and subsequent peripheral pathological neovascularization. Our findings demonstrate also that candesartan induced VEGR2 activation but did not alter the VEGF expression in hypoxia. These effects coincided with restoration of the redox-state balance by reducing the anti-angiogenic and nitrative stress mediator, iNOS, and upregulation of the antioxidant and proangiogenic modulator HO-1. We believe that the higher dose of candesartan (10 mg/kg/day, i.p.) can activate other pathways involved in angiogenesis including AT2 receptor activation as shown recently by our group [7] or HO-1 pathway as illustrated in the current study.
The HO-1 is an enzyme degrading heme into carbon monoxide, free iron and biliverdin. HO-1 is widely distributed and highly induced by a range of stimuli including inflammation, oxidative stress and nitric oxide (reviewed in [26]). Our results showed that retinas from the hypoxic group showed significant reduction in HO-1 expression compared with retinas exposed to normoxia and that candesartan treatment (10 mg/kg/day, i.p.) was able to restore expression of HO-1 under hypoxia. Our results lend further support to previously reported neuro- and vascular protective action of HO-1 induction in models of retinal ischemia [27, 28] and diabetic retina [29].
HO-1 contributes to the formation of blood vessels both directly, through enhancing the angiogenic activities of EC, and indirectly, through regulating VEGF expression in vivo and in vitro [30]. Our results showed that while candesartan treatment (p12–p17) induced expression of HO-1 under normoxia and hypoxia, it tends to increase VEGF but did not reach statistical significance in vivo. Therefore, we further investigated the role of HO-1 in candesartan-mediated VEGF expression and angiogenic response in human retinal EC under both normoxia and hypoxia. Under hypoxia, candesartan did not stimulate VEGF expression compared with hypoxia alone, which mirrors the in vivo results. Moreover, silencing HO-1 expression in EC undergoing hypoxia stimulated VEGF production, which recapitulates the in vivo results where hypoxic retinas were burdened with reduced HO-1 expression and enhanced VEGF expression. Silencing HO-1 expression in EC mitigated candesartan-induced VEGFR2 angiogenic response in normoxia (Figs. 7, 8). In support, our recent work demonstrated that candesartan induced VEGF-angiogenic response in brain EC [11]. Remarkably, silencing HO-1 expression completely blunted candesartan-mediated VEGFR2 activation and alignment of EC into tube-like structures. Our results are in agreement with prior studies that demonstrated requirement of HO-1 to mediate angiogenic response in retinal EC [20, 21]. This finding supports the notion that candesartan-induced HO-1 expression acts as molecular switch to favor VEGFR2-mediated angiogenic signal and enhances reparative angiogenesis in the ischemic retina.
Recent studies suggested inverse relationship between HO-1 and iNOS expression, where restoring HO-1 or carbon monoxide inhibits iNOS expression [31, 32]. There is increasing evidence that iNOS is anti-angiogenic [18]. We and others demonstrated that increases in retinal oxidative and nitrative stress contribute to vaso-obliteration in models of ischemic retinopathy [16, 22, 33–37]. Nitrative stress driven by enhanced iNOS expression in the ischemic retina was postulated as the molecular switch that down-regulates physiological intra-retinal angiogenesis and facilitates pathological angiogenesis toward the vitreous [15, 16, 38]. Our results show that treatment with candesartan during relative hypoxia (p12–p17) completely prevented hypoxia-induced iNOS expression, pathological neovascularization and enhanced revascularization of the central retina (Figs. 1, 2, 3). The protective effect of candesartan was associated with significant decreases in retinal nitrative stress assessed by total nitrite level and nitrotyrosine, the footprint of peroxynitrite (Fig. 3). These results lend further support to prior studies that demonstrated protective effects of ARBs including candesartan [14], losartan, [39], olmesartan [40] and telmisartan [41]. Prior evidence demonstrated the protective effects of ARBs via reducing iNOS expression in various models [42–44]. To our knowledge, this is the first report that links the vascular protective effect of candesartan to modulating iNOS expression in the ischemic retinopathy model.
Astrocytes and Müller glia are essential for regulating the retinal capillaries during development and maintaining barrier properties in maturation (reviewed in [45]). While AT1 and AT2 receptors are well expressed in the various layers of retina, AT1 receptor expression is prominent in glial Muller cells [46]. Our results showed that blocking the AT1 receptor with candesartan significantly inhibited hypoxia-induced activation of Muller cells evident by marked reduction of radial expression of GFAP (Fig. 4b). In agreement, treatment with valsartan prevented Muller activation in an ischemic retinopathy model [47]. Prior studies identified glial Muller cells as a primary site for increased iNOS expression in response to hypoxia [15, 38, 48]. Our results showed that hypoxia induced expression of iNOS in the INL of the central ischemic retina (Fig. 4a) with strong co-localization of iNOS with both Muller glial cells and EC in the deep retina capillaries (Fig. 4; Supplementary Fig. 2). Increases in iNOS expression in the ischemic retina and the adjacent EC conferred inhibition of physiological angiogenesis and facilitated growth of aberrant blood vessels. Together, candesartan stimulated reparative angiogenesis and prevented pathological angiogenesis in ischemic retinopathy. Our results identify a novel mechanism by which candesartan exerts its protective action on the retina vasculature via stimulation of HO-1-dependent VEGFR2 activation and angiogenic response as well as inhibition of iNOS. Candesartan is already approved by the FDA and has a favorable safety profile. Hence, there is great potential to repurpose the drug for the treatment of ischemic retinopathy including both proliferative diabetic retinopathy and retinopathy of prematurity.
Abbreviations
- VEGF:
-
Vascular endothelial growth factor
- p:
-
Postnatal day
- Cand:
-
Candesartan
- ROP:
-
Retinopathy of prematurity
- DR:
-
Diabetic retinopathy
- iNOS:
-
Inducible nitric oxide synthase
- HO-1:
-
Hemeoxygenase-1
References
El-Kenawi AE, El-Remessy AB (2013) Angiogenesis inhibitors in cancer therapy: mechanistic perspective on classification and treatment rationales. Br J Pharmacol 170:712–729
Sapieha P (2012) Eyeing central neurons in vascular growth and reparative angiogenesis. Blood 120:2182–2194
Chen J, Smith LE (2007) Retinopathy of prematurity. Angiogenesis 10:133–140
Carmeliet P (2003) Angiogenesis in health and disease. Nat Med 9:653–660
Fletcher EL, Phipps JA, Ward MM, Vessey KA, Wilkinson-Berka JL (2010) The renin-angiotensin system in retinal health and disease: its influence on neurons, glia and the vasculature. Prog Retin Eye Res 29:284–311
Chua CC, Hamdy RC, Chua BH (1998) Upregulation of vascular endothelial growth factor by angiotensin II in rat heart endothelial cells. Biochim Biophys Acta 1401:187–194
Alhusban A, Kozak A, Ergul A, Fagan SC (2013) AT1 receptor antagonism is proangiogenic in the brain: BDNF a novel mediator. J Pharmacol Exp Ther 344:348–359
Willis LM, El-Remessy AB, Somanath PR, Deremer DL, Fagan SC (2011) Angiotensin receptor blockers and angiogenesis: clinical and experimental evidence. Clin Sci (Lond) 120:307–319
Kozak A, Ergul A, El-Remessy AB, Johnson MH, Machado LS et al (2009) Candesartan augments ischemia-induced proangiogenic state and results in sustained improvement after stroke. Stroke 40:1870–1876
Guan W, Somanath PR, Kozak A, Goc A, El-Remessy AB et al (2011) Vascular protection by angiotensin receptor antagonism involves differential VEGF expression in both hemispheres after experimental stroke. PLoS One 6:e24551
Soliman SA, Ishrat T, Pillai A, Somanath PR, Ergul A et al (2014) candesartan induces a prolonged proangiogenic effect and augments endothelium-mediated neuroprotection after oxygen and glucose deprivation: role of vascular endothelial growth factors A and B. J Pharmacol Exp Ther 349:444–457
Sano H, Hosokawa K, Kidoya H, Takakura N (2006) Negative regulation of VEGF-induced vascular leakage by blockade of angiotensin II type 1 receptor. Arterioscler Thromb Vasc Biol 26:2673–2680
Nagisa Y, Shintani A, Nakagawa S (2001) The angiotensin II receptor antagonist candesartan cilexetil (TCV-116) ameliorates retinal disorders in rats. Diabetologia 44:883–888
Nakamura S, Tsuruma K, Shimazawa M, Hara H (2012) Candesartan, an angiotensin II type 1 receptor antagonist, inhibits pathological retinal neovascularization by downregulating VEGF receptor-2 expression. Eur J Pharmacol 685:8–14
Sennlaub F, Courtois Y, Goureau O (2001) Inducible nitric oxide synthase mediates the change from retinal to vitreal neovascularization in ischemic retinopathy. J Clin Invest 107:717–725
Sennlaub F, Courtois Y, Goureau O (2002) Inducible nitric oxide synthase mediates retinal apoptosis in ischemic proliferative retinopathy. J Neurosci 22:3987–3993
Gu W, Weihrauch D, Tanaka K, Tessmer JP, Pagel PS et al (2003) Reactive oxygen species are critical mediators of coronary collateral development in a canine model. Am J Physiol Heart Circ Physiol 285:H1582–H1589
Zhang Q, Zhang J, Guan Y, Zhang S, Zhu C et al (2009) Suppression of retinal neovascularization by the iNOS inhibitor aminoguanidine in mice of oxygen-induced retinopathy. Graefes Arch Clin Exp Ophthalmol 247:919–927
Du AJ, Ren B, Gao XW, Yang L, Fu Y et al (2013) Effects of aminoguanidine on retinal apoptosis in mice with oxygen-induced retinopathy. Int J Ophthalmol 6:436–441
Xu Z, Gong J, Maiti D, Vong L, Wu L et al (2012) MEF2C ablation in endothelial cells reduces retinal vessel loss and suppresses pathologic retinal neovascularization in oxygen-induced retinopathy. Am J Pathol 180:2548–2560
Zhang W, Zhang X, Lu H, Matsukura M, Zhao J et al (2013) Silencing heme oxygenase-1 gene expression in retinal pigment epithelial cells inhibits proliferation, migration and tube formation of cocultured endothelial cells. Biochem Biophys Res Commun 434:492–497
Abdelsaid MA, Pillai BA, Matragoon S, Prakash R, Al-Shabrawey M et al (2010) Early intervention of tyrosine nitration prevents vaso-obliteration and neovascularization in ischemic retinopathy. J Pharmacol Exp Ther 332:125–134
Abdelsaid MA, Matragoon S, El-Remessy AB (2013) Thioredoxin-interacting protein expression is required for VEGF-mediated angiogenic signal in endothelial cells. Antioxid Redox Signal 19:2199–2212
Wei Y, Gong J, Thimmulappa RK, Kosmider B, Biswal S et al (2013) Nrf2 acts cell-autonomously in endothelium to regulate tip cell formation and vascular branching. Proc Natl Acad Sci USA 110:E3910–E3918
El-Remessy AB, Behzadian MA, Abou-Mohamed G, Franklin T, Caldwell RW et al (2003) Experimental diabetes causes breakdown of the blood-retina barrier by a mechanism involving tyrosine nitration and increases in expression of vascular endothelial growth factor and urokinase plasminogen activator receptor. Am J Pathol 162:1995–2004
Grochot-Przeczek A, Dulak J, Jozkowicz A (2010) Heme oxygenase-1 in neovascularisation: a diabetic perspective. Thromb Haemost 104:424–431
Liu XQ, Wu BJ, Pan WH, Zhang XM, Liu JH et al (2013) Resveratrol mitigates rat retinal ischemic injury: the roles of matrix metalloproteinase-9, inducible nitric oxide, and heme oxygenase-1. J Ocul Pharmacol Ther 29:33–40
Peng PH, Chao HM, Juan SH, Chen CF, Liu JH et al (2011) Pharmacological preconditioning by low dose cobalt protoporphyrin induces heme oxygenase-1 overexpression and alleviates retinal ischemia-reperfusion injury in rats. Curr Eye Res 36:238–246
Fan J, Xu G, Jiang T, Qin Y (2012) Pharmacologic induction of heme oxygenase-1 plays a protective role in diabetic retinopathy in rats. Invest Ophthalmol Vis Sci 53:6541–6556
Bussolati B, Mason JC (2006) Dual role of VEGF-induced heme-oxygenase-1 in angiogenesis. Antioxid Redox Signal 8:1153–1163
Quincozes-Santos A, Bobermin LD, Latini A, Wajner M, Souza DO et al (2013) Resveratrol protects C6 astrocyte cell line against hydrogen peroxide-induced oxidative stress through heme oxygenase 1. PLoS One 8:e64372
Rosales MA, Silva KC, Duarte DA, de Oliveira MG, de Souza GF et al (2014) S-nitrosoglutathione inhibits inducible nitric oxide synthase upregulation by redox posttranslational modification in experimental diabetic retinopathy. Invest Ophthalmol Vis Sci 55:2921–2932
Papp A, Nemeth I, Karg E, Papp E (1999) Glutathione status in retinopathy of prematurity. Free Radic Biol Med 27:738–743
Gu X, El-Remessy AB, Brooks SE, Al-Shabrawey M, Tsai NT et al (2003) Hyperoxia induces retinal vascular endothelial cell apoptosis through formation of peroxynitrite. Am J Physiol Cell Physiol 285:C546–C554
Brooks SE, Gu X, Samuel S, Marcus DM, Bartoli M et al (2001) Reduced severity of oxygen-induced retinopathy in eNOS-deficient mice. Invest Ophthalmol Vis Sci 42:222–228
El-Remessy AB, Al-Shabrawey M, Platt DH, Bartoli M, Behzadian MA et al (2007) Peroxynitrite mediates VEGF’s angiogenic signal and function via a nitration-independent mechanism in endothelial cells. FASEB J 21:2528–2539
Stevenson L, Matesanz N, Colhoun L, Edgar K, Devine A et al (2010) Reduced nitro-oxidative stress and neural cell death suggests a protective role for microglial cells in TNFalpha−/− mice in ischemic retinopathy. Invest Ophthalmol Vis Sci 51:3291–3299
DeNiro M, Al-Halafi A, Al-Mohanna FH, Alsmadi O, Al-Mohanna FA (2010) Pleiotropic effects of YC-1 selectively inhibit pathological retinal neovascularization and promote physiological revascularization in a mouse model of oxygen-induced retinopathy. Mol Pharmacol 77:348–367
Nakamura H, Yamazaki M, Ohyama T, Inoue T, Arakawa N et al (2009) Role of angiotensin II type 1 receptor on retinal vascular leakage in a rat oxygen-induced retinopathy model. Ophthalmic Res 41:210–215
Moravski CJ, Kelly DJ, Cooper ME, Gilbert RE, Bertram JF et al (2000) Retinal neovascularization is prevented by blockade of the renin–angiotensin system. Hypertension 36:1099–1104
Nagai N, Noda K, Urano T, Kubota Y, Shinoda H et al (2005) Selective suppression of pathologic, but not physiologic, retinal neovascularization by blocking the angiotensin II type 1 receptor. Invest Ophthalmol Vis Sci 46:1078–1084
Gemici B, Tan R, Ongut G, Izgut-Uysal VN (2010) Expressions of inducible nitric oxide synthase and cyclooxygenase-2 in gastric ischemia-reperfusion: role of angiotensin II. J Surg Res 161:126–133
Palaniyappan A, Uwiera RR, Idikio H, Menon V, Jugdutt C et al (2013) Attenuation of increased secretory leukocyte protease inhibitor, matricellular proteins and angiotensin II and left ventricular remodeling by candesartan and omapatrilat during healing after reperfused myocardial infarction. Mol Cell Biochem 376:175–188
Fan Q, Liao J, Kobayashi M, Yamashita M, Gu L et al (2004) Candesartan reduced advanced glycation end-products accumulation and diminished nitro-oxidative stress in type 2 diabetic KK/Ta mice. Nephrol Dial Transplant 19:3012–3020
Fletcher EL, Downie LE, Hatzopoulos K, Vessey KA, Ward MM et al (2010) The significance of neuronal and glial cell changes in the rat retina during oxygen-induced retinopathy. Doc Ophthalmol 120:67–86
Kurihara T, Ozawa Y, Shinoda K, Nagai N, Inoue M et al (2006) Neuroprotective effects of angiotensin II type 1 receptor (AT1R) blocker, telmisartan, via modulating AT1R and AT2R signaling in retinal inflammation. Invest Ophthalmol Vis Sci 47:5545–5552
Downie LE, Pianta MJ, Vingrys AJ, Wilkinson-Berka JL, Fletcher EL (2008) AT1 receptor inhibition prevents astrocyte degeneration and restores vascular growth in oxygen-induced retinopathy. Glia 56:1076–1090
Palmer LA, Semenza GL, Stoler MH, Johns RA (1998) Hypoxia induces type II NOS gene expression in pulmonary artery endothelial cells via HIF-1. Am J Physiol 274:L212–L219
Acknowledgments
The authors would like to thank Astra-Zeneca for providing candesartan. Authors are indebted to Megan L. Bartasis for providing help with Western blot. This work was supported by grants from EY-022408, JDRF (2-2008-149) and Culver Vision Discovery Institute to ABE, postdoctoral Fellowship from Islamic Development Bank to M.F.E., predoctoral fellowship (12PRE12030197) from American Heart Association for S.S., VA Merit award (BX000891) to S.C.F.
Conflict of interest
The authors have nothing to disclose. The contents do not represent the views of the Department of Veterans Affairs or the United States government.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Shanab, A.Y., Elshaer, S.L., El-Azab, M.F. et al. Candesartan stimulates reparative angiogenesis in ischemic retinopathy model: role of hemeoxygenase-1 (HO-1). Angiogenesis 18, 137–150 (2015). https://doi.org/10.1007/s10456-014-9451-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10456-014-9451-4