
Abstract
In this work, we report Na-Fe(III) coordination polymer, Na[Fe(C10H12N2O8)(H2O)]·2H2O (1), derived from Ethylenediaminetetraacetic acid (EDTA) and the compound exhibits distorted octahedral coordination geometry. The polymeric structure is generated when octahedral sodium is connected with five oxygens comes from five EDTA molecules and one water molecule. The specific surface area (SBET) of the compound was measured by Brunauer-Emmett-Teller (BET) which is 1036.9653 m²/g that value is consider as impressive. Further, thermal stability, sorption properties of nitrogen, water and benzene were also examined. In addition, electrical conductivity, the amount of ions and Hirshfeld surface analysis were also studied. The N2 adsorption isotherm displays two distinct stages at low relative pressures (P/P0 = 0-0.02), indicating the presence of two distinct types of nanopores within the material structure. In case of adsorption of water molecules, the adsorption capacity, reaching nearly 29 mol/kg, and achieves saturation at water vapor pressure while the benzene primarily adsorbs onto the outer surface of the sorbent material at 1.38 mol/kg but its adsorption capacity is lower in comparsion to water. This observation indicates that delineating the adsorption texture of the adsorbent material. The cleavage of certain bonds between the polymerized metal and the ligand in the coordination compound is confirmed through electrical conductivity and the amount of ions. The Hirshfeld surface analysis provided information about various types of surfaces interactions in this compound.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Over the recent years, porous coordination polymers (PCPs) have garnered significant interest, standing out as particularly captivating and promising subjects of research within various scientific investigations [1]. PCPs are materials distinguished by their expansive network architectures, intricately crafted through the coordination bonds that link metal nodes—comprising both metal ions and clusters—with organic ligands. This assembly results in a complex and extensive framework that defines the unique characteristics of PCPs within the realm of materials science [2,3,4]. In contrast to conventional inorganic porous materials like zeolite molecular sieves and micro-porous silica, PCPs offer distinct advantages in terms of both structural and compositional diversity and stand out for their customizable porosity and ease of functionalization [5]. Within the realm of 2D polymers, 2D coordination polymers (CPs) represent a distinctive subclass of organic materials. They are crafted by systematically arranging organic ligands alongside metal ions, creating a two-dimensional landscape. In the literature, these 2D CPs are frequently denoted by various terms such as 2D metal-organic frameworks (2D MOFs), coordination nanosheets (CONASHs), and metal-organic layers (MOLs) [6]. CPs showcase dimensionality and porosity as prominent features. These characteristics stem from the diverse arrays formed through the repetition of metal-ligand bound units, giving rise to networks that can manifest in one, two, or three dimensions [7]. CPs are regarded as an appealing subject of research owing to their diverse structures and noteworthy applications, particularly in photocatalysis [8,9,10], ion exchange [11], magnetism [12,13,14], and significant contributions to areas such as adsorption, separation, and sensing [15,16,17]. Moreover, CPs exhibit proficiency as effective adsorbents, with the potential for adsorption taking place on surfaces, within encapsulated spaces, in interstices, interiors, or through chemical reactivity [18]. Furthermore, a multitude of CPs showcase notable sorption capacities. For instance, the MOF-177 compound demonstrates the capability to adsorb over 1.3 g of nitrogen at 77 K [19]. The presence of expansive cavities in these frameworks, coupled with the low heats of sorption [20]—characterized by van der Waals sorbent-sorbate interactions—facilitates the achievement of sorption-desorption cycles under standard conditions [21].
Ethylenediaminetetraacetic acid (EDTA) is a tetrabasic acid featuring ten potential donor atoms, comprising eight oxygen atoms and two nitrogen atoms [22]. Further, it has capacity to create up to five-membered metal-N, N and metal-N, O chelate rings underscores its acknowledged capability as a hexadentate chelator with numerous metal ions. Furthermore, the conformational flexibility of the metal-N, N’-ethylenediamine ring appears to contribute to the augmentation of coordination mode diversity [23]. EDTA is widely recognized as an economical and biocompatible organic molecule [24]. Given its numerous N and O coordination sites, coupled with flexible connection modes, it has proven to be an exceptionally intriguing candidate for the construction of metal-organic coordination polymers and complexes. Furthermore, it has been established that coordination compounds based on EDTA play crucial roles in electrochemistry, magnetism, and the detection of toxic metal ions [25,26,27].
We utilized the disodium salt of ethylenediaminetetraacetic acid (EDTA-H2Na2), known for its facile formation of coordination bonds with iron ions. Consequently, a 2D coordination polymer was synthesized, showcasing suitability for applications in the previously mentioned fields. To enhance the formation of the 2D polymer, a surfactant (5-sulfosalicyl acid) was introduced as an additive. This addition significantly contributed to the interaction between the iron ions and the ligand, resulting in the formation of a distinct 2D polymer. The surfactant played a pivotal role in orchestrating a unique polymerization process, leading to the creation of a polymer with a substantial surface area. Comprehensive studies were conducted on the compound, including an examination of its structure, thermal stability, electrical, sorption properties, and Hirshfeld surface area.
2 Experimental
All chemicals used in this study were sourced from Sigma-Aldrich and employed in their original form. The elemental composition, expressed as a percentage, was calculated using the Dumas method with ELEMENTARY UNICUBE® IZI equipment. FT-IR spectrum was recorded on a Bruker Invenio S-2021 spectrometer. The UV spectrophotometric technique, Cary 5000 UV-Vis-NIR instrument, Agilent Technologies, was employed to examine the structure of the compound across a wavelength range of 200–1100 nm. TG/DTA were measured on Simultaneous DTG-60, DTA-TG apparatus from Shimadzu. In this research, we applied low-temperature nitrogen adsorption at 77 K, utilizing a Quantachrome® ASiQwin™- Automated Gas Sorption unit, to analyze and characterize the porous properties of the compound. Nitrogen partial pressure was systematically varied between 0.005 and 0.995 P/P0 throughout the experiments, corresponding to nitrogen adsorption and desorption curves. To process the adsorption curves and extract key parameters such as micropore volume and average pore diameter, we employed well-established analytical methods including the BET (Brunauer-Emmett-Teller), Langmuir, Dollimore and Heale (DH), Dubinin Radushkevich (DR), t-graph, and As-graph methods. To provide a comprehensive characterization of the porous structure, we applied the Barrett-Joyner-Halenda (BJH) method to estimate the volume of mesopores within the samples. For a more detailed analysis, pore volume and size distribution information were obtained using both the Density Functional Theory (DFT) method and the Barrett-Joyner-Halenda (BJH) method. The average pore diameter (Dav), as calculated using the BET formula, is determined by the equation: Dav = 4 V/S, where Dav represents the average pore diameter, V signifies the volume of adsorbed nitrogen, and S corresponds to the surface area. Electrical conductivity and ion concentration were determined using a Bante510-DH Benchtop Conductivity/TDS Meter for the aqueous solution.
2.1 Materials
All chemicals were used without further purification. Iron (III) nitrate nonahydrate (99.5%), disodium salt of ethylenediaminetetraacetic acid dihydrate (Trilon B, 99%), 5-sulfosalicylic acid (99.5%) and distilled water.
2.2 Synthesis
Firstly, the first solution was prepared by dissolving the disodium salt of ethylenediaminetetraacetic acid dihydrate (EDTA-H2Na2·2H2O) (22.32 g, 0.06 mmol) in distilled water (0.3 mol). Subsequently, the second solution was prepared by dissolving Fe(NO3)3 • 9H2O (20.2 g, 0.05 mmol) in distilled water (0.3 mol). Next, the third solution was prepared by dissolving 5-sulphosalicylic acid (C7H6O6S) (20 g, 0.092 mmol) in distilled water (0.3 mol). After preparing the solutions, the first and second solutions were slowly added together, and then the third solution was gradually added to the resulting mixture. After a certain period, a brown solution was formed. Subsequently, the solution was placed in a teflon autoclave at 323 K for 72 h. Following this, the solution was kept in a thermostat at 303 K for 10 days to allow crystal growth. Dark brown cubic crystals were obtained after 3–4 days. Yield: 65% (based on Fe and Na): Anal.calcd. for 1: C10H19N2O11FeNa: C, 28.436%, found: C, 28.112% Found: C, 28%; H, 4.2%; O, 41.3%; N, 6.1%. FTIR (cm− 1): 3380 (br), 3084(m), 1612 (s), 1477(w), 1352 (s), 1244 (s), 1175(s), 1127(m), 1088(m), 1037 (m), 927(w), 878(w), 834(w), 807(w), 719(m), 657(m), 585(m), 567(m), 510(m), 471(m).Scheme 1

Synthesis of 1
2.3 X-ray crystallography
Single XRD measurements was performed on Rigaku Oxford Diffraction, data collection and cell refinement were executed with a sealed CuKα (λ = 0. 1.54184Å) X-ray source on a four-circle diffractometer using HyPix3000 detector. The structure was solved through SHELXTL [28, 29], implemented in OLEX2 [30]. The deposited cif has CCDC No. = 2336258 for 1 and the crystallographic data is depicted in Table S1 (Supplementary Material).
3 Results and discussion
3.1 UV-visible spectral studies
The incidence of ultraviolet (UV) light waves on the aqueous solution of a coordination compound led to the absorption of light at a wavelength of λmax = 478 nm. In this spectral region, the Fe3+ ion caused the electrons in the 3d orbitals to undergo d-d electron transitions, moving from the lower energy d orbitals to the higher energy d orbitals (Fig. 1a).
Moreover, the absorption of UV light with a wavelength of λmax = 309 nm led to ligand-to-metal charge transfer (LMCT) within the coordination compound. This LMCT process involved the transfer of an electron from the highest occupied molecular orbital (HOMO) of the Trilon B ligand to the lowest unoccupied molecular orbital (LUMO) of the Fe3+ ion.
Furthermore, the absorption of light with a wavelength of λmax = 260 nm, which falls in the short-wavelength ultraviolet (UVA) region, caused direct n→π* electronic transitions involving the unshared electron pairs in the C = O groups of the Trilon B molecule within the coordination compound. This electromagnetic light energy pushed the lone pairs of oxygen towards the relaxing orbitals of the π bonds, altering the electronic configuration of the molecule.
Additionally, light waves with a wavelength of λmax = 222 nm led to the transfer of the unshared electron pairs of the oxygen atoms in the molecule to n→σ* transitions, resulting in a different electronic rearrangement within the coordination compound (Fig. 1b).

UV-Vis spectra for 1. (a) d-d transition region for iron ion; (b) ligand-metal charge transfer (LMCT) and specific transition domains in ligands
3.2 X-ray crystallography
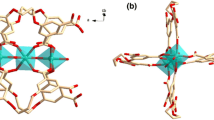
The compound (1) Na[Fe(C10H12N2O8)(H2O)].2H2O is composed of iron(III) with distorted octahedral coordination (Fig. 2). The iron is bonded with the two nitrogens and the three oxygens of the EDTA and a molecule of water to complete the sphere. One -COO− is connected with the Fe from one oxygen and to a sodium from the other. The sodium is octahedral coordinated with five oxygens from 5 EDTA molecules and one water molecule to make a polymeric structure. The distances and angles around the Fe are, for Fe-O: Fe1-O3 = 1.977(2) Å, Fe1-O5 = 2.122(2) Å, Fe1-O6 = 1.969 (2) Å and Fe1-O7 = 2.099(2) Å, and for Fe-N: Fe1-N1 = 2.319(2) Å nd Fe1-N2 = 2.318 (2) Å. The long distances Fe1-O5 is for Fe-OH2, the long distances in case of Fe-N can be explained by the high spin state in Fe. These bond distances are in agreement with other published data for complexes with EDTA [31,32,33]. Porosity data for 1 showed a void of 1513.37 ų (96.34%) per unit cell (Fig. 3). A search of the Cambridge Structural Database indicated that López-Alcalá, et al. [31] and Altaba et al. [33] reported same compound as discrete complex but in fact it is coordination polymer. The selected bond distances, angles and H-bonds are listed in Tables S2 and S3 (Supplementary Material), respectively.

Labelled ball-and-stick of 1

Representation of porosity of 1
3.3 TG and DTA analysis
The initial step involved holding the sample at 25 °C under argon, flow rate of 80 ml/min for 10 min and afterward temperature was increased at a rate of 10 °C/min, spanning from 25 °C to 800 °C. The analysis revealed that the compound experienced a total mass loss of approximately 67.567%. The remaining 32.433% primarily consists of iron (III) oxide, sodium oxide, and residual graphite within the compound (Figure S1, Supplementary Material). The TG and DTA curves exhibited a close correspondence, mutually reinforcing each other. Within the temperature range of 30 °C to 800 °C, mass loss occurred predominantly in two distinct regions on the graph. The initial region was observed within the temperature range of 30 °C to 407.43 °C. Considering the hydrophilic nature of the compound, this initial loss can be attributed to the evaporation of a minimal amount of absorbed gases and moisture present within the compound. The endothermic process is elucidated by a DTA peak at 97 °C, indicating an activation energy of 56.13 J/g. Within this range, a mass decrease of 46.554% (theoretically 45.97%) was observed, corresponding to the desorption of crystalline hydrate H2O molecules within the pores. As the temperature increases, the structure of the compound is gradually compromised, ultimately leading to the disintegration of carboxyl groups within the ligand. This process is exothermic, reaching its maximum DTA peak at 308.64 °C, and is concluded with an activation energy of 791.55 J/g. The second region is characterized by temperatures above 407.43 °C, where 21.013% (theoretically 22.043%) of the weight loss is attributed to the remaining ligand. This process is also exothermic, with the polymer structure undergoing its most significant transformation, as indicated by the highest DTA peak at 471.64 °C. The thermal stability of the compound at 600 °C was re-examined because there was almost no change between 600 °C and 800 °C. The obtained residue was analyzed using PXRD, and the results indicated that the final product consists of iron and carbon (Figure S2, Supplementary Material).
3.4 Adsorption property of the coordination polymer
The examination of nitrogen adsorption and the determination of textural characteristics are actively pursued fields of scientific research. This is particularly relevant in the context of developing new materials with enhanced properties. Textural attributes such as surface area, porosity, and pore size play pivotal roles in shaping the functional properties of materials. They significantly influence adsorption capacity, activity, and stability. In recent times, there has been significant interest in the domain of coordination polymer, especially those involving the combination of iron (III) nitrate and EDTA-H2Na2. These combinations stand out due to their unique chemical and structural compositions. Metal-organic polymers have high porosity, therefore, the property of absorbing or separating gases is considered important [34,35,36,37]. The BET surface area of these compounds with different spatial dimensions differs sharply from each other. For example, the average surface for one-dimensional (1D) compounds is 250–550 m2/g [34, 38] average surface area for two-dimensional (2D) compounds is 300–1200 m2/g and for three-dimensional (3D) compounds the average surface area is 1000–10,000 m2/g comes. As can be seen from the above data, the morphology of metal organic polymers has a great influence on its porosity [39]. Such compounds hold the potential to exhibit remarkable adsorption properties, particularly concerning nitrogen adsorption. Investigating the nitrogen adsorption behavior on this compound not only deepens our understanding of its textural and structural attributes but also uncovers potential applications, particularly in pharmaceutical domains. Figure 4 presents the N2 adsorption characteristics for the synthesized material. The analysis reveals a significant capacity for N2 adsorption. The adsorption isotherm displays two distinct stages at low relative pressures (P/P0 = 0-0.02), indicating the presence of two distinct types of nanopores within the material’s structure. Subsequently, there is a further increase in the volume of adsorbed nitrogen, extending to values P/P0 = 0-0.02. Table S4 (Supplementary Material), in conjunction with Figure S3 (Supplementary Material), provides critical parameters such as surface area, pore sizes, and their volume distribution. These parameters were determined using appropriate adsorption isotherm models. These findings significantly contribute to our understanding of the material’s textural characteristics and have substantial implications for potential applications.

Nitrogen adsorption isotherms on 1
Table S4, Supplementary Material, presents vital information regarding the textural characteristics of the porous material [C10H19FeN2NaO11]n, highlighting its outstanding surface properties. Specifically, the specific surface area (SBET) measures an impressive 1036.9653 m²/g, a value of immense importance for these materials. This extensive surface area has the potential to enhance bonding with active compounds or other molecules, providing notable advantages across various applications. Remarkably, the presence of micropores, with a micropore area of 263.48 m²/g, underscores the material’s efficiency in adsorbing small molecules. Conversely, the external surface area of the material, excluding micropores, may play a role in adsorbing larger molecules or facilitating fluid transport. The determination of mesopore volume using the Barrett-Joyner-Halenda (BJH) method yields a value of 1.525 cm³/g. This underscores the predominantly microporous nature of the material, characterized by a higher volume of micropores compared to mesopores. The maximum pore volume (HK) is recorded as 7.551467 cm³/g, indicating the substantial capacity of the material to accommodate adsorbed substances. Additionally, the mean hydraulic pore radius, as determined by the MP (Macropore) method, measures 10.4423 Å. These textural properties hold particular significance for pharmaceutical and various other applications, as they can significantly influence the material’s adsorption capacity, dissolution rate, bioavailability, and other critical attributes. Materials with high surface area and pore volume may exhibit enhanced adsorption of active compounds, making them potentially valuable in controlled-release formulations and various applications.
Figure S3, Supplementary Material, demonstrates a comprehensive depiction of the differential distribution of pores based on their respective diameters within the material under analysis. This systematic investigation has revealed the presence of two distinct categories of pores within the material: Micropores (S-pores): These micropores possess diameters that fall within the range of 1.1 to 1.9 nanometers. Mesopores: Mesopores are discerned with diameters that span from approximately 2 to 20 nanometers. An intriguing feature of this distribution is the marked augmentation in the abundance of S-pores as one progresses from smaller to larger diameters within the interval of 1.1–1.9 nanometers. Moreover, for pores bearing diameters of 2–20 nanometers, the recorded volume is estimated to be approximately 0.0084 cubic centimeters per gram (cm³/g). Nevertheless, upon examination of pores with diameters surpassing 2 nanometers, a consistent reduction in their prevalence is observed. Their volume within the size range of 7.0-15.1 nanometers is calculated to be approximately 0.0095 cm³/g. As the pore diameter continues to increase, their presence on the material’s surface gradually diminishes. The relative volume ratio of L-pores to S-pores within the analyzed sample is a matter of notable significance. This ratio spans a range from 1 nm to 17.7–20 nanometers, thereby highlighting the multifaceted distribution and proportion of pores inherent to the material. These insights into the distribution of pore sizes assume pivotal importance as they greatly contribute to our comprehension of the material’s adsorption properties and, consequently, have substantial implications for its potential applications across a myriad of fields.
The investigation of adsorption isotherms pertaining to water and benzene vapors on CP (1) was conducted under controlled conditions within a high-vacuum McBen-Bakr balance, maintaining room temperature as the standard. This instrumental setup is distinguished by its inclusion of a quartz coil, boasting an impressive sensitivity level of 1.78 × 10^-3 kg/m. The determination of the specific surface area (S) of the sorbent was achieved by meticulous utilization of the BET theory equation, which relies on the structural parameters of adsorption. From the resultant adsorption isotherms concerning benzene and water vapors, crucial parameters, including the monolayer capacity (αm), saturation volume (Vs), and their respective relative surface areas (S), were ascertained. These determinations provide critical insights into the behavior of the sorbent material. Figure 5 serves as a graphical representation of the isotherm elucidating the adsorption of water on the compound. The vertical axis is dedicated to the measurement of adsorbed water, denoted in mol/kg, while the horizontal axis correlates with the relative pressure of saturated water vapor under standard room temperature conditions. Upon meticulous scrutiny of the data embedded within the adsorption isotherms, it becomes apparent that the adsorbent material exhibits a distinct affinity for water molecules. Notably, it manifests an impressive adsorption capacity, reaching nearly 29 mol/kg, and achieves saturation at water vapor pressure.

Water adsorption isotherms on 1
The hydrophilic nature of the sorbent is evident, and the occurrence of capillary condensation phenomena is minimal. The active centers that reside within the adsorption pores of each cell of the adsorbent material actively and participate indirectly in the adsorption of water molecules. Adsorbates engage with these active centers, culminating in the formation of ion-molecular aqua complexes. The classification of the adsorption isotherm for this sample aligns with type III, as per the established categorization proposed by Brunauer. The markedly high adsorption coefficient observed within the relative pressure range, spanning from the initial saturation pressure of the adsorbate to its ultimate saturation pressure, underscores the substantial specific surface area and the significant pore volume, as graphically represented in Fig. 5. The data acquired during these adsorption processes substantiate prior findings regarding the heterogeneous composition and structural attributes of the synthesized polymer. Moreover, these findings reaffirm the surface properties intrinsic to the adsorbent material. Within this context, it is noteworthy that water molecules predominantly adsorb onto active centers featuring oxygen, iron, and nitrogen elements. Shifting the focus to the adsorption isotherms pertaining to benzene, an intriguing observation arises concerning the rapid escalation in adsorption values, even at low relative pressures, up to R/Rs ≈ 0.1. Subsequently, the adsorption converges gradually toward saturation, displaying an undulating pattern. The steep ascent in adsorption isotherms at such low relative pressures (up to Р/Рs ≈ 0.1) implies the presence of a substantial adsorption potential at the initial saturation point.
Benzene molecules, upon initial adsorption, form T- and π-complexes, assuming a sandwich-like configuration with benzene on the outer surface of the adsorbent material, and their bond strength is partially guaranteed. Given that benzene primarily adsorbs onto the outer surface of the sorbent material, its adsorption capacity is notably lower in comparsion to water, measuring at 1.381 mol/kg ( Fig. 6). This observation is further corroborated by indicators delineating the adsorption texture of the adsorbent material.

Benzene adsorption isotherms on 1
3.5 Hirshfeld surface analysis
The Hirshfeld surface analysis of 1 was performed using CrystalExplorer 21.5 software [40]. This software generated the Hirshfeld surfaces, which provide valuable insights into the intermolecular interactions within the compounds. To determine the normalized contact distance (dnorm) in the analysis of Hirshfeld surfaces, the following equation was utilized:
If di in this equation represents the distance from the surface to the nearest atom inside, the van der Waals (vdW) radius of this atom can be determined by ri. Similarly, de in the equation represents the distance to the nearest atom outside the surface, and re represents the van der Waals radius of this atom. The Hirshfeld surface 2D fingerprint plot provides a visualization of the contribution of all atoms and interatomic interactions to the formation of the surface.
The Hirshfeld surface analysis of 1 was conducted on its asymmetric unit, excluding the outer space water molecule from the analysis. A normalized contact distance (dnorm) surface was generated and depicted using a color scheme that represents atomic dimensions ranging from − 0.6858 to 1.5850. In the resulting picture, regions colored in red, white, and blue indicate different types of Hirshfeld surface contacts with fragments located within a distance of up to 3.8 Å from 1 (Fig. 7).
The Hirshfeld’s total surface volume was determined to be 357.52 ų, while the total surface area measured 313.73 Ų. These values provide quantitative information about the extent and characteristics of the surface interactions present in 1.

A dnorm representation of a Hirshfeld surface. Hirshfeld interaction of the surface with fragments up to a distance of 3.8 Å
During the formation of the Hirshfeld surface, the di values were determined to range from 0.6825 to 2.8725 Å, while the de values ranged from 0.6864 to 2.6076 Å. In this case, the fact that di > de indicates that the Hirshfeld volume is relatively large compared to the level.
The contribution of both internal and external atoms to the formation of the Hirshfeld surface can be observed in the 2D fingerprint images of the surface (Fig. 8). According to these images, the primary contribution to the surface area arises from O‧‧‧H/H‧‧‧O interactions, accounting for 54.8% of the total. H‧‧‧H interactions contribute 27.7%, Na‧‧‧O interactions contribute 11%, H‧‧‧C interactions contribute 2.3%, O‧‧‧O interactions contribute 1.9%, O‧‧‧C interactions contribute 1.2%, and Na‧‧‧H interactions contribute 1.1%. These interatomic interactions collectively shape the characteristics of the Hirshfeld surface.
Furthermore, the contributions of elements in compound-1 to the formation of the surface can be analyzed. Among the total contributions, All-Na accounts for 5.8%, All-O accounts for 32.2%, All-H accounts for 60.6%, and All-C accounts for 1.4%. Conversely, Na-All contributes 6.4%, O-All contributes 38.5%, H-All contributes 52.9%, and C-All contributes 2.2% to the overall interatomic interactions within the Hirshfeld surface.

A 2D fingerprint image of a Hirshfeld surface showing the percentage contribution of interatomic interactions to the surface
3.6 Electrical conductivity and the amount of ions
Initially, the compound was dissolved in water, yielding three distinct outcomes during the experiment. The temperature was incrementally raised from 20 °C to 90 °C, resulting in a reduction of electrical conductivity from 4.29 mS to 4.03 mS and a decrease in ion concentration from 2.14 ppt to 2.04 ppt until the temperature reached 55 °C. Subsequently, as the temperature increased to 60 °C, both electrical conductivity and ion concentration began to rise. At 90 °C, electrical conductivity reached 5 mS, and the ion concentration equaled 2.54 ppt. This observation can be attributed to the cleavage of certain bonds between the polymerized metal and the ligand in the coordination compound (Fig. 9).
Following this, the solution was cooled from 90 °C to 20 °C. As a result, when the temperature was lowered to 25 °C, electrical conductivity increased until 7.24 mS, and the ion concentration rose until 3.6 ppt. However, when the temperature was further decreased to 20 °C, both electrical conductivity and ion concentration decreased (Fig. 10).

The electrical conductivity and ion content of the two-dimensional iron coordination polymer change as temperature increases

The electrical conductivity and ion content of the two-dimensional iron coordination polymer vary as temperature decreases
4 Conclusion
Na-Fe(III) based coordination polymer has been prepared under hydrothermal conditions and characterized by different spectroscopic techniques, TGA, elemental analysis and X-rays diffraction method. The remarkable SBET 1036.9653 m²/g of the compound has been identified and explored this polymer for variety of applications such as sorption of nitrogen, water and benzene. The N2 adsorption isotherm demonstrates about the nanopores within the material structure. Further, water molecules indicate the good adsorption capacity (23 mol/kg) in comparison to the to the benzene (0.849 mol/kg) that describe about the adsorption texture of the adsorbent material. These findings contribute prominently about the textural characteristics and their potential applications particularly in pharmaceuticals. The electrical conductivity investigation provides information about the cleavage of certain bonds between the polymerized metal and the ligand. Hirshfeld surface analysis indicated that total surface volume (357.52 ų) and total surface area (313.73 Ų) provide quantitative information about the extent and characteristics of the surface interactions exist in compound.
Data availability
All relevant data are available in this manuscript or its supplementary material.
References
Tang, L.-P., Yang, S., Liu, D., Wang, C., Ge, Y., Tang, L.-M., Zhou, R.-L.: J. Mater. Chem. A. 8, 14356–14383 (2020)
O’Keeffe, M., Yaghi, O.M.: Chem. Rev. 112, 675–702 (2012)
Zhou, H.-C., Long, J.R., Yaghi, O.M.: Chem. Rev. 112, 673–674 (2012)
Schoedel, A., Li, M., Li, D., O’Keeffe, M., Yaghi, O.M.: Chem. Rev. 116, 12466–12535 (2016)
Long, J.R., Yaghi, O.M.: Chem. Soc. Rev. 38, 1213–1214 (2009)
Tran, M., Kline, K., Qin, Y., Shen, Y., Green, M.D., Tongay, S.: Appl. Phys. Rev. 6, 041311 (2019)
Batten, S.R., Champness, N.R., Chen, X.-M., Garcia-Martinez, J., Kitagawa, S., Öhrström, L., O’Keeffe, M., Suh, M.P.: J. Reedijk CrystEngComm. 14, 3001–3004 (2012)
Zhu, H., Liu, D., Zou, D., Zhang, J.: J. Mater. Chem. A. 6, 6130–6154 (2018)
Zhang, H., Liu, G., Shi, L., Liu, H., Wang, T., Ye, J.: Nano Energy. 22, 149–168 (2016)
Waseem, S., Saleem, H., Akhtar, M.N., Imran, M., Javaid, A., AlDamen, M.A., Bikas, R., Khanfar, M.A.: Inorg. Chim. Acta. 559, 121756 (2024)
Liu, J., Zheng, M., Wu, S., Zhang, L.: Coord. Chem. Rev. 483, 215084 (2023)
Akhtar, M.N., Tahir, S., Sadakiyo, M., Warsi, M.F., AlDamen, M.A., Song, Y.: Transit. Met. Chem. 42, 635–641 (2017)
Akhtar, M.N., Mateen, M., Sadakiyo, M., Warsi, M.F., AlDamen, M.A., Song, Y.: J. Mol. Str. 1141, 170–175 (2017)
Akhtar, M.N., Chen, Y.-C., AlDamen, M.A., Tong, M.-L.: Dalton Trans. 46, 116–124 (2017)
Jaros, S.W., Sokolnicki, J., Wołoszyn, A., Haukka, M., Kirillov, A.M., Smoleński, P.: J. Mater. Chem. C. 6, 1670–1678 (2018)
Huang, W., Jiang, J., Wu, D., Xu, J., Xue, B., Kirillov, A.M.: Inorg. Chem. 54, 10524–10526 (2015)
Hasan, Z., Jhung, S.H.: J. Hazard. Mater. 283, 329–339 (2015)
Baruah, J.B.: Coord. Chem. Rev. 470, 214694–214723 (2022)
Rowsell, J.L.C., Yaghi, O.M.: Microporous Mesoporous Mater. 73, 3–14 (2004)
Chowdhury, P., Bikkina, C., Gumma, S.: J. Phys. Chem. C. 113, 6616–6621 (2009)
Khabibjonovich, A.A., Yusupboevich, Y.Y., Khamidjanovich, R.A., Sadullayevich, N.A., Bakhtiyarovich, I.A., Akhtar, M.N., AlDamen, M.A., Mengnorovich, A.J.: J. Mol. Str. 1295, 136581 (2024)
Liu, D.-S., Qiu, Z.-J., Fu, X., Liu, Y.-Z., Ding, P., Zhu, Y.-X., Sui, Y.: J. Solid State Chem. 278, 120879 (2019)
Belmont-Sánchez, J.C., García-Rubiño, M.E., Frontera, A., Matilla-Hernández, A., Castiñeiras, A.: Niclós-Gutiérrez, J.; Crystals 10, 391 (2020)
Janicki, R., Mondry, A.: Dalton Trans. 48, 3380–3391 (2019)
Chatterjee, D., Oszajca, M., Katafias, A., van Eldik, R.: Coord. Chem. Rev. 436, 213773 (2021)
Holmberg, R.J., Korobkov, I., Murugesu, M.: RSC Adv. 6, 72510–72518 (2016)
Zhang, K., Dai, Z., Zhang, W., Gao, Q., Dai, Y., Xia, F., Zhang, X.: Coord. Chem. Rev. 434, 213809 (2021)
Sheldrick, G.M.: Acta Crystallogr Sect. A: Found. Crystallogr. 71, 3–8 (2015)
Sheldrick, G.M.: Acta Crystallogr Sect. A: Found. Crystallogr. 64, 112–122 (2008)
Dolomanov, O.V., Bourhis, L.J., Gildea, R.J., Howard, J.A.K., Puschmann, H.: J. Appl. Cryst. 42, 339–341 (2009)
By, J.M., López-Alcalá, M.C., Puerta-Vizcaíno, Gonzalez-Vílchez, F.: Acta Cryst. C40, 939–941 (1984)
Kocot, P., Karocki, A., Stasicka, Z.: J. Photochem. Photobiol A: Chem. 179, 176–183 (2006)
By, X., Solans, Altaba, M.F.: Acta Cryst. C40, 635–638 (1984)
Farha, O.K., Spokoyny, A.M., Mulfort, K.L., Galli, S., Hupp, J.T., Mirkin, C.A. Small. 5, 1727–1731 (2009).
Li, T.-T., Liu, Y.-M., Wang, T., Wu, Y.-L., He, Y.-L., Yang, R., Zheng, S.-R. Microporous Mesoporous Mater. 272, 101–108 (2018).
Mauger-Sonnek, K., Streicher, L.K., Lamp, O.P., Ellern, A., Weeks, C.L. Inorg. Chim. Acta. 418, 73–83 (2014).
Khabibjonovich, A.A., Yusupboevich, Y.Y., Khamidjanovich, R.A., Sadullayevich, N.A., Bakhtiyarovich, I.A., Akhtar, M.N., AlDamen, M.A. Mengnorovich. J. Mol. Struct. 1295, 136581 (2024).
Polunin, R.A., Kolotilov, S.V., Kiskin, M.A., Cador, O., Golhen, S., Shvets, O.V., Ouahab, L., Dobrokhotova, Z.V., Ovcharenko, V.I., Eremenko, I.L., Novotortsev, V.M., Pavlishchuk, V.V. Eur. J. Inorg. Chem. 2011, 4985–4992 (2011).
Furukawa, H., Cordova, K.E., O’Keeffe, M., Yaghi, O.M.: 341, 1230444 (2013).
Spackman, P.R., Turner, M.J., McKinnon, J.J., Wolff, S.K., Grimwood, D.J., Jayatilaka, D., Spackman, M.A.: J. Appl. Crystallogr. 54, 1006–1011 (2021)
Acknowledgements
The authors would like to thank the Uzbekistan government for direct financial support of the research.
Funding
M.N. Akhtar is grateful to Higher Education Commission of Pakistan for research grant (project number = 20-15570).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Ethical approval
Not applicable.
Consent to participate
Informed consent was obtained from all individual participants included in the study.
Consent to publication
All authors are agreed to submit this manuscript.
Conflict of interest
The authors declare no competing financial interest
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Khabibjonovich, A.A., Yusupboyevich, Y.Y., Zamirovich, A.B. et al. A coordination polymer of Na-Fe(III) with EDTA: synthesis, characterization, electrical conductivity, adsorption properties and Hirshfeld surface analysis. Adsorption 30, 1407–1418 (2024). https://doi.org/10.1007/s10450-024-00504-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10450-024-00504-4