
Abstract
This paper describes the design, functioning and use of a portable detection platform for multiplex nucleic acid testing. The system features a bead-supported DNA hybridization assay performed inside a microfluidic cartridge. Polystyrene particles modified with DNA capture probes are confined in the detection area and exposed to a solution of fluorescently labeled target DNA strands. The cartridge, fabricated from inexpensive thermoplastic polymers, allows for conducting up to eight assays in parallel. The detection instrument is equipped with a pneumatic module and a manifold lid serving as an interface to mediate fluid displacement on the cartridge. The fluorescence signal deriving from each assay is recorded by a semi-confocal fluorescence reader embedded in the detection platform. The compact design of the instrument and its level of integration make it possible to obtain an analytical result in less than 15 min, while only few manual steps need to be performed in between. A proof-of-concept demonstration involving Cy3-labeled, PCR-amplified genomic DNA confirms the ability to detect Bacillus anthracis in a multiplexed single-assay format using lef and capC genes. Limits of quantification are on the order of 1 × 109 copies/μL for lef targets.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Rapid detection of pathogenic agents is crucial for public health and life stock management in the event of a bioterrorist attack (Inglesby et al. 1999; Lim et al. 2005). The ability to not only confirm (or disprove) the presence of a biological hazard but also accurately identify its phenotype at a contaminated site would enable authorities to implement adequate countermeasures with minimum delay. Despite significant advances in the development of suitable detection methods, on-site testing remains challenging, however, and first-responder teams are often limited to performing risk-assessment while collected samples are transferred to a secured environment for in-depth investigation (Emanuel et al. 2008). One reason is the diversity of pathogenic agents that can range from toxins to virus and bacteria, each of which requires a different detection approach. Another reason is the incompatibility of many technologies with unfavorable conditions in the field that include humidity, shock and temperature-variations, among others. A third reason is the constitution of the sample which often contains low levels of microbial content in a complex matrix (e.g., air, soil, water or food), demanding for purification and concentration of target analytes—a process that is both time intensive and labor intensive (Lim et al. 2005; Isabel et al. 2012; Geissler et al. 2012).
Detection of bacterial species typically involves culture enrichment and selection, while identification relies on nucleic acid testing (NAT). NAT takes advantage of the specificity provided by recombinant base pairs in DNA strands. Polymerase chain reaction (PCR) has become a standard technique to this end given its ability to produce multiple copies of a target sequence, making it possible to detect low abundant genes with high sensitivity in a short period of time (Persing and Tenover 2011). However, PCR is difficult to perform in a field setting using standard instrumentation. Microarray-based technologies allow for screening of multiple target strands simultaneously, but the use of common (two-dimensional) arrays involves a stochastic hybridization process under non-equilibrium conditions, often resulting in prolonged analysis times. Micro total analysis systems (μTAS) hold the potential for increased analytical throughput while reducing sample consumption, manual intervention and associated risks of contamination. The capacity of miniaturized, integrated systems to perform complex analytical procedures has been demonstrated in a number of ways (Madou et al. 2006; Mark et al. 2010; Meltzer et al. 2011; Neethirajan et al. 2011). Still, their potential for facilitating real-life tasks has not materialized yet.
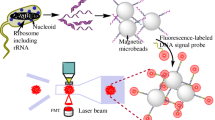
Herein, we describe our efforts in developing a μTAS platform that complies with many—though not all—of the requirements for biothreat detection. We conceived a robust, compact instrument to ensure safe portability and autonomous operation outside a standard laboratory setting. The system uses a disposable microfluidic cartridge to perform multiplex NAT assays, which involve packing of beads, modified with capture probes, inside a detection area to form a solid-phase extraction matrix for nucleic acids in a target analyte solution (Fig. 1). Fluidic operations on the cartridge are mediated by a pneumatic module, while read-out of the assay is achieved through an integrated optical detection system. We perform assay and instrument development using NAT targets that are produced by PCR amplification and fluorescent labeling of genomic DNA eventually extracted from a contaminated matrix (e.g., powder or soil). The possibility of performing multiple assays along with the respective controls using a single fluorophore simplifies both the design of the instrument and the analytical protocol. Ultimately, integration of sample preparation steps as our preceding developments for on-site sample isolation (Isabel et al. 2012) and DNA extraction (Geissler et al. 2012) along with PCR amplification or the implementation of a label-free detection step (Brouard et al. 2011) would considerably increase versatility and effectiveness of the instrument presented herein.

Simplified flow chart (left-hand panel) detailing the different steps to be performed on the cartridge along with instrumental implementations (right-hand panel) enabling operation in a semi-automated fashion
Fluorescence-based detection schemes are widely used for μTAS since they provide sensitivity and potentially high spatial resolution, though their practical implementation remains challenging: Rayleigh or Raman scattering of excitation light by microfluidic features, intrinsic fluorescence from polymer-based materials used for device fabrication and the presence of concomitant species in the sample matrix can severely affect the signal-to-background ratio and thus the detection limit achievable by the assay. Interfering effects can be reduced by confining the target analyte in a small volume for which the use of microbeads provides a plausible solution. To this end, fluorescence measurements with particle-supported assays have mainly been performed using camera-based detection methods (Zaytseva et al. 2005; de Jong and Lucy 2006; Wen et al. 2008; Schonbrun et al. 2010)—an approach that is usually less sensitive and more expensive than using single-channel detectors. Furthermore, the use of cameras often requires intensive image processing and data analysis that can be detrimental to rapid testing. We devised a non-imaging, semi-confocal fluorescence detection system (Fig. 2a) that provides improved sensitivity and measurement speed while promoting convenient integration in our μTAS platform.

Configuration of the detection system. a Simplified schematic of the optical setup (cross-sectional view). The pathways for excitation and fluorescence light are depicted in blue and green, respectively. b Design of the fiber-coupled LED-based excitation source. c Design of the integrated semi-confocal detection head. Only the collected fluorescence beam path is shown (in green) for clarity (color figure online)
The use of bead-based assays is advantageous since particles provide larger surface areas than inner channel walls for target capture. Tailoring the surface chemistry for the immobilization of capture probes is also more convenient to do on particles (off-chip) than within the fluidic system itself. Bead-based assays typically involve magnetic separation of particle-bound target analyte from a sample solution using permanent magnets (Jiang and Harrison 2000; Zaytseva et al. 2005). Dubus et al. (2006) have demonstrated the use of microfabricated electromagnets for on-chip detection of DNA strands on microbeads using confocal fluorescence imaging. Bead-based capture of analyte molecules has further been shown with non-magnetic particles using weirs or constrictions embedded in the microfluidic circuit (Seong and Crooks 2002; Ali et al. 2003; Kim et al. 2006; Shin et al. 2007; Zhang et al. 2007, 2008). Herein, we adopted this capture strategy due to its simplicity in design and ease of integration as compared to active magnetic confinement. Bead beds have been shown to enhance mixing and reaction rates for biomolecular processes such as DNA hybridization (Seong and Crooks 2002; Kim et al. 2006). While the formation of both monolayer (Zhang et al. 2007, 2008) and multilayer configurations (Kim et al. 2006; Shin et al. 2007) has been reported, systematic investigation on the benefits of one particular approach for optical detection is yet to be achieved. The work presented herein compares the impact of bead packing strategies (i.e., monolayer vs. multilayer) on the fluorescence signal repeatability using a multichannel chip design.
We focus on Bacillus anthracis to demonstrate the possibility of detecting the presence of a biothreat agent using the instrumentation developed in this work. Bacillus anthracis is classified as a high-priority pathogen considering the severity of illness it can inflict (Inglesby et al. 1999). The virulence of B. anthracis is associated with the presence of two plasmids: pXO1 that contains genes for the production of toxins (Robertson et al. 1988; Bragg and Robertson 1989) and pXO2 that contains genes responsible for capsule biosynthesis (Green et al. 1985; Uchida et al. 1985; Makino et al. 1989). Strains lacking either plasmid are non-virulent or exhibit attenuated virulence (Uchida et al. 1986). However, similar genes can be present in non-pathogenic Bacillus strains (Luna et al. 2006), making it necessary to detect more than one target sequence, ideally on both plasmids, to identify virulent strains of B. anthracis in a reliable fashion. We selected two unique sequence targets for this study—the lef gene encoding the lethal factor (located on pXO1) and the capC gene implicated in capsule biosynthesis (located on pXO2). In addition, a sequence from Streptococcus agalactiae was included as a negative control.
2 Experimental section
2.1 PCR amplification
Primers and probes (Table 1) were designed with sequences available in local or NCBI databases using GCG and Oligo6 software (Molecular Biology Insights, Cascade, CO). Streptococcus agalactiae primers were adapted from previous work (Picard et al. 2009). Single-stranded, Cy3-labeled PCR amplicons were prepared according to published procedures (Boissinot et al. 2007; Parham et al. 2007) using Cy3-labeled primers to generate the labeled target strand, and phosphorylated primers to generate the complementary strand. Purified genomic DNA (1,000 copies) from B. anthracis (Ames strain; provided by the Public Health Agency of Canada, National Microbiology Laboratory, Winnipeg, MB) was amplified in a PCR mixture containing 10 mM Tris–HCl (pH 9.1; Sigma-Aldrich, Oakville, ON), 50 mM KCl (Sigma-Aldrich), 2.5 mM MgCl2 (Sigma-Aldrich), 3.3 g/L bovine serum albumin (BSA; Sigma-Aldrich), 0.1 % Triton X-100 (Sigma-Aldrich), 0.2 mM dNTPs (Invitrogen, Burlington, ON), 0.025 U/μL Taq DNA polymerase (Thermo Fisher Scientific, Waltham, MA) coupled to TaqStart antibody (Clontech Laboratories, Mountain View, CA) and four primers (Integrated DNA Technologies, Coralville, IA) at 0.4 μM each. The volume of the PCR mixture was 50 μL. Negative controls were produced using 1 ng (4.1 × 106 copies) of genomic DNA from S. agalactiae ATCC-13813 and a primer pair targeting a portion of the cfb gene (Picard et al. 2009). Amplification was done in a PTC-200 thermocycler (Bio-Rad Laboratories, Hercules, CA) using the following conditions: 3 min at 94 °C, then n cycles (where n = 30, 35 or 40) of 1 s at 95 °C followed by 30 s at 55 °C for B. anthracis (56 °C for S. agalactiae) and finally a 2-min extension step at 72 °C. Once amplification was completed, the concentration of the amplified sample was evaluated on agarose gel using a DNA mass ladder from Invitrogen (Table 2) and an AlphaImager system (Alpha Innotech Corp., Santa Clara, CA). Concentration values were calculated on the basis of an average mass of 660 Da and an amplicon length of 239 bp for capC and 180 bp for lef. The final concentration for each amplicon was divided by two to take into account the digestion of the unlabeled complementary strand, which was realized by adding 8 μL containing 10 units of lambda exonuclease (New England Biolabs, Ipswich, MA) to the PCR mixture followed by incubation at 37 °C for 5 min (Boissinot et al. 2007). The exonuclease was then inactivated by heat treatment at 75 °C for 5 min. The resulting digested amplicon solution was fractionated and stored at 4 °C in 0.6-mL centrifuge microtubes for a maximum of 1 week. Prior to hybridization experiments, digested amplicons were diluted 1:4 in hybridization buffer to a final volume of 200 μL and filtered on a low-binding membrane (0.1-μm pore diameter; Millipore, Billerica, MA) using centrifugation at 12,000×g for 2 min.
2.2 Fabrication of the microfluidic cartridge
Fluidic structures were imprinted into Versaflex CL30 (GLS Corp., McHenry, IL) using a mold prepared by photolithography employing SU-8 resist on a 6″ silicon wafer (Silicon Quest International, Santa Clara, CA). The wafer was heated on a hot plate at 200 °C for 10 min. SU-8 (Gersteltec, Pully, Switzerland) was applied on the wafer by spin coating. Following a pre-bake (65 °C for 5 min, then 95 °C for 15 min using a temperature ramp of 2 °C/min), the wafer was exposed to UV light (365 nm, Hg i-line) through a transparency photomask (Fine Line Imaging, Colorado Springs, CO) using a Model 6200 mask aligner (EV Group, Schärding, Austria). Post-exposure bake was done using the same conditions as for the pre-bake. Resist features were developed in propylene glycol monomethyl ether acetate (PGMEA; Sigma-Aldrich) for 1–2 min (depending on thickness), followed by rinsing with PGMEA and isopropanol (Anachemia Science, Lachine, QC) and drying with a stream of nitrogen gas. The master was hard-baked at 130 °C for 2 h and finally coated with a thin, anti-adhesive layer formed from 1H, 1H, 2H, 2H-perfluorooctyl-trichlorosilane (Sigma-Aldrich) using deposition from the vapor phase under reduced pressure. Versaflex CL30 was received in the form of pellets and was extruded at 165 °C to yield a 2-mm-thick film which was cut in pieces of 6″ in diameter. Imprinting was done using an EVG 520 embossing tool (EV Group) operated at 135 °C, a pressure of ~1 × 10−3 mbar, and a force of 1.2 × 103 N applied over the entire area of the wafer. The patterned elastomer was peeled off the master before cooling to room temperature and stored for at least 12 h before further processing. Openings defining reservoirs were punched manually.
Zeonor 1060R (Zeon Chemicals, Louisville, KY) was obtained in the form of pellets. The material was shaped to disks (6″ in diameter) using an Engel 150 injection molding apparatus operated at temperatures of between 127 and 134 °C, an injection speed of 45–103 mm/s and a pressure of 25 bar. The mold (stainless steel, custom-fabricated) was cooled for 15 s before the disk was released. Access holes were drilled with a 2.8-mm-diameter drill bit using a computerized milling machine. The stack was assembled manually and remained untouched for ~12 h to ensure proper adhesion between the plastic layers. The cartridge was released as a rectangular cutout section (72 mm in width, 112 mm in length and 4 mm in thickness) using a high-pressure water jet (performed by Transformation Eclipse, Boucherville, QC). Throughout the fabrication process, top and bottom parts of the cartridge were protected using adhesive tape (Semiconductor Equipment Corp., Moorpark, CA) to prevent damage to the plastic surfaces. All fabrication steps were carried out in a class 1,000 clean room environment.
2.3 Pneumatic system
High-pressure nitrogen gas cylinders (2,700 psi) were acquired from Leland Ltd. (South Plainfield, NJ). A puncture device (Leland) mediates the flux of compressed gas to an internal pressure regulator (Leland) to provide an output of 90 psi. A second regulator (Parker Hannifin Corp., Cleveland, OH) controlled by a DC set-point voltage (0–5 V) is used to adjust the pressure between 0 and 30 psi (1 % accuracy). Gas supply lines upstream of the second regulator are made of stainless steel (1/8″ in diameter; Swagelok Quebec Group, Saint-Laurent, QC). A pressure port distributes the regulated output through eight individual three-way control valves (The Lee Company, Westbrook, CT), each one connected to a designated exit on the manifold lid using Tygon R-3603 vacuum tubing (1/8″ in diameter). A one-touch fitting (SMC Corp. of America, Noblesville, IN) is used to connect the system to a standard pressure line. A three-way tap (Swagelok) is used to switch from one pressure source to the other. The frame supporting the manifold lid features a screw-based lock system that holds the interface lid in place once contact with the cartridge is established. The manifold lid (85 mm in width, 112 in length and 13 mm in thickness) was machined from a block of Plexiglas and entailed a set of through-holes to mediate transfer of pressure to the cartridge. Each pressure outlet was confined with a rubber-elastic O-ring to provide an air-tight seal. The interface lid is enclosed with a light-protecting ABS cap to shield the detector and prevent interference with ambient light during detection experiments.
2.4 Optical detection system
A high-power LED emitting at 530 nm (245 mW, Green Rebel, Luxeon Star LEDs; Quadica Developments, Brantford, ON) operated using a controllable DC driver (BuckPuck, 700 mA maximum, Luxeon Star LEDs; Quadica) was used as a light source (Fig. 2b). The light was sent through a band pass interference filter (FF01-530/11; Semrock, Rochester, NY) to shape the excitation spectral profile and eliminate cross talk with the fluorescence detection channel. The filtered light was focused using a molded concentrator (Carclofibre coupling lens, 20 mm, Luxeon Star LEDs; Quadica) and was coupled into a multimode optical fiber (core diameter: 300 μm, NA = 0.37; Thorlabs, Newton, NJ) terminated with an adjustable collimator at the fiber end (F-H10-VIS-FC; Newport Instruments Canada Corp., Mississauga, ON). A core diameter of 300 μm was chosen as the best compromise in terms of coupling efficiency versus exclusion of unwanted modes in the intensity profile. The excitation beam from the fiber-coupled LED was steered by a dichroic beam splitter (LPD01-532RS; Semrock) and sent to the sample located at the focal plane of a microscope objective (4×, NA = 0.13; Olympus, Richmond Hill, ON) (Fig. 2c). An excitation power of 18 μW was delivered at the focal plane. The excitation beam at this location had a circular, 400-μm-diameter, top hat profile. A miniature PC-board CMOS placed on the transmission side of the dichroic filter allows the bead packing process to be monitored during the assay development stages using off-axis illumination with a ring of red-emitting LEDs (λ > 650 nm) positioned around the objective. Fluorescence emitted from the sample was collected by the same objective lens and steered by a second dichroic beam splitter (FF650-Di01; Semrock) toward an interference filter (FF01-575/25; Semrock) and focused into a multimode optical fiber (core diameter: 200 μm, NA = 0.22; OZ Optics, Carp, ON) using a plano-convex lens (50-mm focal length; Thorlabs). The core aperture of the light collection fiber plays the role of a classical confocal pinhole, rejecting light that originates from outside the region of interest. The fiber diameter was selected for optimal collection of fluorescence light from a statistically sufficient number of beads (~80) while avoiding the collection of out-of-focus light (scatter and auto-fluorescence of plastic materials) as well as light coming from outside the fluidic channels. The light collection fiber output was connected to a photon-counting PMT module (H7421-40; Hamamatsu Corp., Bridgewater, NJ). Time-integrated pulse counts were captured using custom-made circuitry based on a microprocessor module (RabbitCore RCM4300 Series; Digi International, Minnetonka, MN) and displayed on a 4.3″ LCD touch screen located on the front panel of the instrument. The setup was packaged in a custom-built sheet metal case (Protocase, Sydney, NS) to ensure convenient operation and handling of the instrument.
2.5 Preparation of beads
Streptavidin-coated polystyrene particles with a nominal diameter (d nominal) of 20 μm were purchased as a suspension (0.5 % w/v, ~9 × 105 beads/mL) from Spherotech (Lake Forest, IL). Suspensions of SuperAvidin-coated microspheres (d nominal = 10 μm, 1 % w/v, 1.57 × 107 beads/mL) and fluorescent microspheres (Envy Green fluorophore, d nominal = 10 μm, 1 % solid fraction) were obtained from Bangs Laboratories (Fishers, IN). The commercial suspension (e.g., of 20-μm beads) was vortexed for 20 s to avoid inaccuracies due to gravitational settling. A volume of 100 μL was transferred into a sterile 0.6-mL centrifuge tube and centrifuged at 20,000×g for 1 min followed by the removal of 70 μL of supernatant. The beads were washed three times with 100 μL of TTL buffer (100 mM Tris–HCl, pH 8.0, 0.1 % Tween 20, 1 M LiCl; Sigma-Aldrich) using vortex agitation for 30 s followed by centrifugation for 1 min at 20,000×g. Beads were finally suspended in 120 μL of TTL buffer to achieve a volume of 150 μL and a concentration of 6 × 102 beads/μL.
To modify the beads with DNA, the suspension was vortexed for 20 s, sonicated for 1 min (to reduce bead aggregation) and vortexed again for 20 s before a 50-μL aliquot (6 × 102 beads/μL) was transferred to a sterile tube and mixed with 75 μL of a 1 μM solution of biotinylated probe oligonucleotide (Integrated DNA Technologies) and 50 μL of TTL buffer. The mixture was vortexed for 5 s and incubated for 10 min at 30 °C and 500 rpm on a Thermomixer R (Eppendorf Canada, Mississauga, ON). The modified beads were centrifuged at 20,000×g for 1 min, followed by removal of 160 μL of the supernatant, washed with 100 μL of 0.15 M NaOH to remove any residual oligonucleotide molecules, vortexed for 30 s and centrifuged again for 1 min at 20,000×g. This washing procedure was repeated once with TT buffer (100 mM Tris–HCl, pH 8.0, 0.1 % Tween 20) and twice with 0.1 % Tween-20. Finally, the beads were suspended in 500 μL of ultrapure (deionized) water (18.2 MΩcm) to a final volume of 515 μL yielding a concentration of ~60 beads/μL. The probe-grafted beads were used within a few hours following their preparation. However, suspending the microbeads in 500 μL of 0.1 % Tween-20 instead of water extends their shelf life to several days under adequate storage and handling conditions. The binding capacity of the microbeads was not determined experimentally. Considering a sixfold excess of probes in solution with respect to the binding capacity provided by the supplier and the incubation time used for bead grafting, it can be assumed that the probe density on the beads does not exceed the optimal density for maximum DNA hybridization efficiency (Kim et al. 2006).
2.6 Formation of bead beds and DNA hybridization
In a typical experiment, 40-μL aliquots of probe-grafted bead suspension (d nominal = 20 μm, adjusted to a concentration of ~60 beads/μL) were pipetted into the injection port of the microfluidic cartridge. The interface lid was closed, and the system was pressurized (7–8 psi) until the entire sample volume was eluted through the channel (typically 2–5 min). The formation of the bead bed was followed on the display in real time. Once completed, the interface lid was opened and 20-μL sample aliquots (corresponding to 30, 35 and 40 PCR cycles as well as a negative control) were eluted and pushed through the cartridge. Hybridization was performed in 8X SSPE buffer containing 0.04 % PVP and 40 % formamide (all purchased from EMD Chemicals, Gibbstown, NJ). Bead beds were rinsed with ultrapure water, and their fluorescence was recorded in a sequential fashion.
3 Results and discussion
3.1 Integrated detection platform
The semi-confocal fluorescence detection scheme developed in this work enables capture of fluorescence from several beads (e.g., ~80 for 20-μm beads) while limiting both the field of view and depth of field to optimize the signal-to-background ratio (Fig. 2). By controlling the excitation beam divergence, the beam footprint at the focal plane can be adjusted to illuminate the entire volume of each detection area. The overlapping excitation and collection waists of this optical design offer a high level of tolerance with respect to the positioning of the fluidic device relative to the objective. Although controlling the divergence of the excitation beam is quite straightforward when using a well-collimated laser source, it is less trivial to achieve when more divergent LED sources are employed. LED beam shaping approaches generally involve a trade-off between beam quality and optical power: A well-collimated beam (that can be focused to a small spot) requires multiple beam shaping elements that considerably reduce the available optical power. A non-corrected (or partially corrected) beam will provide more optical power but will be poorly focused (de Jong and Lucy 2005, 2006). Nevertheless, LEDs offer significant advantages over a laser source in terms of compactness, output power stability, source lifetime and cost (Novak et al. 2007). The present optical setup uses the aperture of a multimode optical fiber as a spatial filter to remove the unwanted light emitted at high angles by the LED and provide a fiber-coupled source with a well-defined spatial profile and numerical aperture while promoting convenient packaging into the instrument housing (see Sect. 2.4 for details).
We conceived a compact and robust design in which optical and pneumatic components are embedded in a custom-designed aluminum housing (Fig. 3a) to ensure safe portability and operation. The instrument can accommodate a high-pressure nitrogen gas cylinder (Fig. 3b) for deployment in a remote environment. The system is equally equipped with a one-touch fitting to connect an external pressure line (e.g., for assay development or testing in a standard laboratory). An internal pressure regulator (Fig. 3c) provides the desired output pressure (e.g., 7–8 psi) from either source (see Sect. 2.3 for details). Injecting sample into the cartridge is done using a micropipette (Fig. 3d) and requires opening and closing of the manifold lid. Alignment marks are used to position the cartridge before the manifold lid is closed. The footprint of the optical interrogation beam is adjusted to illuminate a circular section of the bead bed located in the constriction zone, without the need to scan either the sample or the optics during signal acquisition (Boudreau et al. 2011). An LCD touch screen located at the front panel of the instrument guides the user through the process (Fig. 3e) and allows for convenient intervention when necessary. Pressure is applied to or released from each entry port according to a number coding related to its position on the cartridge (e.g., 1–8). A set of navigation icons allow for accurate displacement of the detection head in both x- and y-directions to perform read-out of the bead beds. Fluorescence data are typically displayed as absolute values that can be used for quantification. It is equally possible to qualify data relative to a set of pre-determined signal intensities as either positive (in the case of hybridization between complementary DNA strands) or negative samples (in the case of non-complementary strands or nucleotide polymorphism in their sequences). A video screen adjacent to the user interface allows visualization of the bead bed during the detection process. Data analysis can be performed at the instrument itself or through a LabView interface on an external computer connected to a USB port.

Photographs illustrating the design and configuration of the integrated detection platform. a Instrument in a standard laboratory setting. b Pneumatic module (exterior). c Pneumatic module (interior part). d Manifold lid in the open position during charging of the cartridge. e Operator defining experimental settings through the touch screen interface. Assignment: (1) cylinder holder (with internal adapter and puncture device), (2) line for compressed nitrogen gas, (3) manifold lid, (4) touch screen LCD, (5) video display, (6) adapter for external pressure line, (7) gas cylinder, (8) tubing for pressure output (to be connected to the manifold lid), (9) pressure regulator (for course control), (10) switching valve, (11) valve holder; (12) pressure regulator (for fine adjustment), (13) lock screw (for closing the manifold lid), (14) pressure outlets with O-ring array, (15) optical port for detection and (16) microfluidic cartridge
3.2 Microfluidic cartridge
The microfluidic cartridge was fabricated from thermoplastic polymers in the form of a three-layer stack with rectangular geometry (Fig. 4a, b). Its central element is a sheet of commercially available styrenic block co-polymer (e.g., VersaflexCL30) which supports the fluidic structures. The thermoelastic properties of this material make it suitable for rapid prototyping based on molding or embossing techniques (Geissler et al. 2009; Roy et al. 2011; Brassard et al. 2011).When processed within the high-temperature regime, the polymer network softens, allowing material to flow and adapt to the master pattern (Roy et al. 2011). Subsequent cooling solidifies the elastomer, thereby preserving the shape of the imprinted features with high accuracy. Top and bottom layers of the cartridge are made of a hard thermoplastic material (e.g., Zeonor 1060R) to provide mechanical stability. The Versaflex sheet serves as a double-sided adhesive that holds the stack together (Fig. 4c). Bonding between Zeonor and Versaflex develops over the course of several hours without mediation by solvents, elevated temperature or pressure, enabling safe, leak-proof manipulation of fluids beyond the specifications provided herein. Zeonor and Versaflex are both optically transparent over a broad range of wavelengths and exhibit relatively low intrinsic fluorescence.

Layout of the microfluidic cartridge used within this study. a The fabrication process involves a set of plastic slides which are superimposed and held together through conformal adhesion (see text for details). The inset illustrates the design of the constriction for which the two levels of depth are encoded in red and blue. b Photograph of an assembled cartridge. c Close-up view of a corner section revealing the three-layer hybrid structure. d Optical micrograph (top view) of the channel inlet with support posts. e Optical micrograph (top view) of the constriction area. As a feature with low aspect ratio, the weir between the channel segments comprises a set of supporting line structures to limit deformation. The ruler beside the channel denotes increments of 100 μm and is used to indicate the length of the bead bed during (or after) formation. f Close-up view of the waste reservoir containing a filter unit to protect the vent. Scale bars correspond to 2 mm, 500, 200 μm and 5 mm for c–f, respectively (color figure online)
The layout comprises a set of eight channels providing an embedded circuitry at the inner bottom of the cartridge. Each channel can be accessed from a macroscopic entry port which can accommodate up to 50 μL of liquid. The gradually converging channel inlets promote a smooth, unhampered approach of microbeads during migration and include a set of support posts (Fig. 4d) to prevent unwanted roof collapse. Microfluidic channels with a width (W) of 200 μm are arranged in parallel with a periodicity of 2 mm when approaching the detection area. A constriction with reduced channel depth (D r) is implemented in each trajectory (Fig. 4e). The outlet of each channel is connected to a macroscopic waste reservoir for collecting sample liquid. A vent allows for air to escape, equilibrating pressure within the conduit. It is possible to mask the vent with a filter membrane (Fig. 4f) that helps prevent escape of excess liquid or aerosol accumulating in the reservoir.
3.3 Bead packing
The beads are stacked against a weir that confines the passage of flow within the fluidic trajectory (Fig. 5a). Precise control of the bead packing is critical given the fact that the semi-confocal system probes the entire depth (D) of the microfluidic channel. We hence optimized D to promote the formation of uniform, monolayer bead beds for particles that are 10 or 20 μm in diameter. Monolayer packing of 20-μm beads (Fig. 5b) was obtained when D and D r were 27 and 12 μm, respectively (Table 3). However, elastomers are prone to deformation and channel dimensions can change as a result of pressure-driven flow (Holden et al. 2003), affecting bead bed characteristics to a large extent (Kim et al. 2006). We typically applied a pressure of 7–8 psi which yielded uniform bead beds while providing reasonable flow-through times. Increasing the pressure compromised regularity in the bead packing due to channel bulging (although the process is reversible and bead beds can re-arrange over time once pressure is released). Lower pressures, on the other hand, slowed the bead bed formation and yielded lengthy analysis times. Bead capture efficiency was estimated at 10–15 %. For example, we typically found 300–400 beads accumulated at the constriction when 40-μL aliquots with a concentration of ~60 beads/μL were injected in the cartridge (Sect. 2.6). In principle, the bead content can diminish at each step of the preparation procedure (Sect. 2.5). We also noticed that beads readily sediment upon injection in the entry port, compromising their displacement within the microfluidic conduit. This limitation can be overcome by implementing an airstream pathway in the manifold to agitate the liquid phase and re-suspend particles prior to the capture process (Geissler et al. 2011).

Bead packing within the microfluidic cartridge. a Schematic illustration of bead confinement using a weir. b–d Optical micrographs of selected bead bed configurations. b Monolayer of 20-μm beads. b Partially irregular packing of 10-μm beads, revealing a monolayer at the periphery and a higher density in the center. c Hybrid stacking comprising a monolayer of 20-μm beads (right-hand side) serving as a plug for a subsequent multilayer of 10-μm beads (left-hand side). Scale bars correspond to 200 μm. e Photograph showing a monolayer bead bed of 20-μm beads as it appears on the video display of the detection platform. The circle indicates the area where fluorescence read-out is performed by the instrument. f Plot of the channel-to-channel variability in fluorescence (6 % RSD) for monolayer bead beds comprising 20-μm particles functionalized with lef oligonucleotide capture probes
We evaluated the impact of bead bed homogeneity on the signal variability from one channel to another (Table 3). The relative standard deviation (RSD) was calculated from the average fluorescence signal recorded for at least three different bead beds. The fluorescence signal was measured over a period of 30 s under stationary conditions (i.e., no flow). RSD on the background signal (RSDBG) using blank beads (i.e., beads without any fluorophore) was relatively invariant, suggesting that channel-to-channel fluorescence signal fluctuations are not correlated to bead size, bead trapping layouts or channel dimensions, suggesting scattering and auto-fluorescence as the principal cause. Uniform bead beds produced with labeled 20-μm beads resulted in channel-to-channel fluctuations of 10 % or less. An increase in D from 27 to 32 μm causes departure from a single-layer bed to an irregular arrangement which augments channel-to-channel signal fluctuations up to 50 % RSD (Table 3). Uniform packing was challenging to achieve with 10-μm beads since the shallow channel (D = 12 μm) is prone to deformation, leading to higher density of beads at the center (Fig. 5c). For comparison, we prepared a multilayer of 10-μm beads in a cartridge with D = 27 μm by first injecting a small amount of the larger beads to retain the smaller ones (Fig. 5d). Here, we recorded channel-to-channel fluorescence signal fluctuations of 10 % RSD or less (Table 3) for the multilayer area, suggesting an overall uniform packing in the detection volume.
The presence of multiple bead layers in the detection volume enhances the signal intensity and, therefore, the assay sensitivity. However, analytical figures of merit will only improve when controlling the bead packing. The formation of multiple layers of 22.7-μm beads did not lead to satisfactory results, accounting for fluctuations of 33 % RSD for a packing in 2–3 layers. While we employed different fluorophores and labeling methods (i.e., surface grafting vs. internal dying), the findings presented in Table 3 suggest that the excitation wavelength, the type of fluorophore and the method of labeling had little or no effect on signal variability. From a practical point of view, 20-μm beads are preferable over 10-μm beads since the microfluidic features can be made larger, which enhances fluid flow and reduces analysis time. We therefore selected 20-μm particles for demonstrating the DNA hybridization assay. To provide a reliable reference for this system, we recorded the intrinsic fluorescence signal of 24 monolayer bead beds formed from lef-modified 22.7-μm beads. The integrity of each bead bed was confirmed on the video display (Fig. 5e) while performing read-out on the same channel segment. The data presented in Fig. 5f account for a channel-to-channel variability of 6 % RSD, suggesting a high degree of reproducibility in both bead packing and average optical response which is conditional for reliable and sensitive detection of hybridization assays.
3.4 NAT assay and detection of B. anthracis
Figure 6a shows typical real-time fluorescence signal traces recorded for a 40-cycle PCR-positive sample (lef) and a negative control (cfb gene of S. agalactiae) injected into separate channels, each containing lef probe beads. The measurement starts with an empty channel into which lef probe-conjugated beads are pumped and packed at the constriction. The signal increases sharply in both channels at the beginning of the hybridization phase due to the presence of Cy3-labeled amplicons in the detection volume. When a positive sample is injected, amplicons bind to probe-conjugated beads, resulting in a positive slope of the fluorescence signal. The negative control, on the other hand, exhibits a constant signal (i.e., the slope equals zero), indicating that no or little hybridization occurs. In a final step, the bead bed is rinsed with water for 5 min to remove non-hybridized amplicons, during which the fluorescence signal stabilizes (Kim et al. 2006). For a positive sample, the fluorescence signal increases significantly while it returns to background level for a negative sample.

DNA hybridization performed on monolayer bead beds comprising 20-μm particles. a Evolution of the recorded fluorescence signal as a function of assay time for a typical detection process involving a positive sample (Cy3-labeled lef amplicon) and a negative control (Cy3-labeled cfb amplicon from S. agalactiae). In this example, 20-μm beads were functionalized with lef oligonucleotide capture probes. The sample comprises 20 μL of a PCR mixture that has undergone 40 cycles of PCR amplification. b Plot of the average fluorescence intensity (background-corrected) for on-bead hybridization and detection of Cy3-labeled amplicons. Target analyte solutions were produced through 30, 35 and 40 cycles of PCR. Error bars represent standard deviations calculated from 3 replicates. c Calibration plot (average fluorescence signal vs. number of DNA copies) for on-bead hybridization and detection of Cy3-labeled amplicons of the lef sequence after 30, 35 and 40 PCR amplification cycles. The error bars represent standard deviations calculated from 3 replicates
The relatively constant slope of the signal trace during hybridization suggests that completion is not reached even for the most concentrated positives samples (i.e., 40 cycles of PCR). This finding can be attributed to the fact that DNA probes are present in excess. Longer durations might therefore lead to more efficient hybridization and hence better sensitivity. The result obtained with the negative control (S. agalactiae) is interesting considering the significant amount of Cy3-labeled amplicons in the samples (1.7 × 10−12 mol or 1 × 1012 copies for a 40-cycle sample aliquot), thereby providing an extreme scenario for testing the specificity of the assay. The measured fluorescence signal during rinsing shows minimal decay over a 5-min period, indicating that photo-induced bleaching of the Cy3 label was negligible during the experiment.
Figure 6b shows the results obtained for the detection of lef and capC target sequences deriving from multiplex PCR amplification. Both B. anthracis target genes can be detected after 30 PCR cycles when the initial sample contains 1,000 copies of genomic DNA. We calculated limits of detection (LOD3σ) of 1 × 109 and 4 × 108 copies/μL for capC and lef, respectively. The higher LOD for the capC assay can be explained by the lower PCR yield for this sequence (as indicated in Table 2). However, both assays exhibit comparable sensitivity when the fluorescence signal is expressed as a function of Cy3-labeled DNA concentration (data not shown). We calculated limits of quantification (LOQ10σ) of 5 × 109 copies/μL for capC and 1 × 109 copies/μL for lef, which makes the declaration of a positive sample statistically valid at 30 cycles of amplification for both targets. Signal variability was higher for lef than for capC hybridization. Differences in flow-through times (e.g., due to variation of the bead bed lengths) are probably the reason for this finding. In addition, hybridization efficiency and dynamics are unlikely to be identical for the two targets (Carletti et al. 2006), which further contributes to the discrepancy observed for lef and capC samples. To account for such variability in hybridization signals, the instrument was designed with the option to provide a simple pass or fail answer (e.g., for rapid testing in a field setting). Since the fluorescence signal shows linear dependency on the concentration of a particular target DNA strand (Fig. 6c), it is possible to establish calibration standards that would facilitate NAT with unspecified sample content.
4 Conclusion
We presented a bead-supported DNA hybridization assay for the selective detection of B. anthracis using a microfluidic cartridge and a stand-alone, prototype instrument. Uniform, single-layer packing of beads was essential to achieve low variability from one assay to another when using a non-imaging approach for fluorescence detection. Virulent strains of B. anthracis can be identified through the use of lef and capC genes labeled with a fluorescent dye. A sequence from S. agalactiae, included as a negative control, confirms excellent selectivity of the B. anthracis hybridization assay. Positive samples can be confirmed after 30 cycles of amplification, corresponding to 5 × 109copies/μL for capC and 1 × 109copies/μL for lef targets. The detection process (with up to 8 assays performed in parallel) can be completed within 15 min, which is appealing in terms of analytical throughput. Optimization of the analytical protocol may, nevertheless, further reduce processing times. The detection scheme, together with the bead-supported assay presented in this study, emphasizes the advantage of simultaneously concentrating particles and decreasing the volume to be probed—a concept that holds promise for detecting minute amounts of a target analyte in complex samples. The instrumental setup may also enable the study of hybridization dynamics since it allows for monitoring fluorescence in real time. Furthermore, integration of sample preparation steps and PCR amplification are options that would make the instrument amenable to field testing. Ultimately, the implementation of a label-free detection scheme (Ho et al. 2002; Brouard et al. 2011) circumventing the need for PCR amplification and fluorescence tagging of nucleic acids would greatly enhance the potential of the μTAS platform in performing point-of-care analysis.
References
Ali MF, Kirby R, Goodey AP, Rodriguez MD, Ellington AD, Neikirk DP, McDevitt JT (2003) DNA hybridization and discrimination of single-nucleotide mismatches using chip-based microbead arrays. Anal Chem 75:4732–4739
Boissinot K, Huletsky A, Peytavi R, Turcotte S, Veillette V, Boissinot M, Picard FJ, Martel EA, Bergeron MG (2007) Rapid exonuclease digestion of PCR-amplified targets for improved microarray hybridization. Clin Chem 53:2020–2023
Boudreau D, Gravel J-F, Voisin B, Le Drogoff B, Veres T (2011) Method and apparatus for detecting fluorescence emitted by particle-bound fluorophores confined by particle traps. US 2011(0226962):A1
Bragg TS, Robertson DL (1989) Nucleotide sequence and analysis of the lethal factor gene (lef) from Bacillus anthracis. Gene 81:45–54
Brassard D, Clime L, Li K, Geissler M, Miville-Godin C, Roy E, Veres T (2011) 3D microfluidic device for biological probe immobilization. Lab Chip 11:4099–4107
Brouard D, Viger ML, Bracamonte AG, Boudreau D (2011) Label-free biosensing based on multilayer fluorescent nanocomposites and a cationic polymeric transducer. ACS Nano 5:1888–1896
Carletti E, Guerra E, Alberti S (2006) The forgotten variables of DNA array hybridization. Trends Biotechnol 24:443–448
de Jong EP, Lucy CA (2005) Spectral filtering of light-emitting diodes for fluorescence detection. Anal Chim Acta 546:37–45
de Jong EP, Lucy CA (2006) Low-picomolar limits of detection using high-power light-emitting diodes for fluorescence. Analyst 131:664–669
Dubus S, Gravel J-F, Le Drogoff B, Nobert P, Veres T, Boudreau D (2006) PCR-free DNA detection using a magnetic bead-supported polymeric transducer and microelectromagnetic traps. Anal Chem 78:4457–4464
Emanuel P, Roos JW, Niyogi K (2008) Sampling for biological agents in the environment. ASM Press, Washington, DC
Geissler M, Roy E, Diaz-Quijada GA, Galas J-C, Veres T (2009) Microfluidic patterning of miniaturized DNA arrays on plastic substrates. ACS Appl Mater Interfaces 1:1387–1395
Geissler M, Voisin B, Veres T (2011) Air stream-mediated vortex agitation of microlitre entities on a fluidic chip. Lab Chip 11:1717–1720
Geissler M, Isabel S, Voisin B, Fauvel C, Boissinot M, Bergeron MG, Veres T (2012) Modular ultrasonic lysis system for rapid nucleic acid extraction and sample transfer of Bacillus spores. J Bioterr Biodef 3:119-1–119-6
Green BD, Battisti L, Koehler TM, Thorne CB, Ivins BE (1985) Demonstration of a capsule plasmid in Bacillus anthracis. Infect Immun 49:291–297
Ho H-A, Boissinot M, Bergeron MG, Corbeil G, Doré K, Boudreau D, Leclerc M (2002) Colorimetric and fluorometric detection of nucleic acids using cationic polythiophene derivatives. Angew Chem Int Ed 41:1548–1551
Holden MA, Kumar S, Beskok A, Cremer PS (2003) Microfluidic diffusion diluter: bulging of PDMS microchannels under pressure-driven flow. J Micromech Microeng 13:412–418
Inglesby TV, Henderson DA, Bartlett JG, Ascher MS, Eitzen E, Friedlander AM, Hauer J, McDade J, Osterholm MT, O’Toole T, Parker G, Perl TM, Russell PK, Tonat K, Working Group on Civilian Biodefense (1999) Anthrax as a biological weapon: Medical and public health management. J Am Med Assoc 281:1735–1745
Isabel S, Boissinot M, Charlebois I, Fauvel CM, Shi L-E, Lévesque J-C, Paquin AT, Bastien M, Stewart G, Leblanc E, Sato S, Bergeron MG (2012) Rapid filtration separation-based sample preparation method for Bacillus spores in powdery and environmental matrices. Appl Environ Microbiol 78:1505–1512
Jiang GF, Harrison DJ (2000) mRNA isolation in a microfluidic device for eventual integration of cDNA library construction. Analyst 125:2176–2179
Kim J, Heo J, Crooks RM (2006) Hybridization of DNA to bead-immobilized probes confined within a microfluidic channel. Langmuir 22:10130–10134
Lim DV, Simpson JM, Kearns EA, Kramer MF (2005) Current and developing technologies for monitoring agents of bioterrorism and biowarfare. Clin Microbiol Rev 18:583–607
Luna VA, King DS, Peak KK, Reeves F, Heberlein-Larson L, Veguilla W, Heller L, Duncan KE, Cannons AC, Amuso P, Cattani J (2006) Bacillus anthracis virulent plasmid pX02 genes found in large plasmids of two other Bacillus species. J Clin Microbiol 44:2367–2377
Madou M, Zoval J, Jia G, Kido H (2006) Lab on a CD. Annu Rev Biomed Eng 8:601–628
Makino S-I, Uchida I, Terakado N, Sasakawa C, Yoshikawa M (1989) Molecular characterization and molecular analysis of the cap region, which is essential for encapsulation of Bacillus anthracis. J Bacteriol 171:722–730
Mark D, Haeberle S, Roth G, von Stetten F, Zengerle R (2010) Microfluidic lab-on-a-chip platforms: requirements, characteristics and applications. Chem Soc Rev 39:1153–1182
Meltzer RH, Krogmeier JR, Kwok LW, Allen R, Crane B, Griffis JW, Knaian L, Kojanian N, Malkin G, Nahas MK, Papkov V, Shaikh S, Vyavahare K, Zhong Q, Zhou Y, Larson JW, Gilmanshin R (2011) A lab-on-chip for biothreat detection using single-molecule DNA mapping. Lab Chip 11:863–873
Neethirajan S, Kobayashi I, Nakajima M, Wu D, Nandagopal S, Lin F (2011) Microfluidics for food, agriculture and biosystems industries. Lab Chip 11:1574–1586
Novak L, Neuzil P, Pipper J, Zhang Y, Lee S (2007) An integrated fluorescence detection system for lab-on-a-chip applications. Lab Chip 7:27–29
Parham NJ, Picard FJ, Peytavi R, Gagnon M, Seyrig G, Gagné P-A, Boissinot M, Bergeron MG (2007) Specific magnetic bead-based capture of genomic DNA from clinical samples: application to the detection of group B streptococci in vaginal/anal swabs. Clin Chem 53:1570–1576
Persing DH, Tenover FC (2011) Molecular microbiology: diagnostic principles and practice (2nd edn). ASM Press, Washington, DC
Picard FJ, Gagnon M, Bernier MR, Parham NJ, Bastien M, Boissinot M, Peytavi R, Bergeron MG (2009) Internal control for nucleic acid testing based on the use of purified Bacillus atrophaeus subsp. globigii spores. J Clin Microbiol 47:751–757
Robertson DL, Tippetts MT, Leppla SH (1988) Nucleotide sequence of the Bacillus anthracis edema factor gene (cya): a calmodulin-dependent adenylate cyclase. Gene 73:363–371
Roy E, Geissler M, Galas J-C, Veres T (2011) Prototyping of microfluidic systems using a commercial thermoplastic elastomer. Microfluid Nanofluid 11:235–244
Schonbrun E, Abate AR, Steinvurzel PE, Weiz DA, Crozier KB (2010) High-throughput fluorescence detection using an integrated zone-plate array. Lab Chip 10:852–856
Seong GH, Crooks RM (2002) Efficient mixing and reactions within microfluidic channels using microbead-supported catalysts. J Am Chem Soc 124:13360–13361
Shin K-S, Lee SW, Han K-C, Kim SK, Yang EK, Park JH, Ju B-K, Kang JY, Kim TS (2007) Amplification of fluorescence with packed beads to enhance the sensitivity of miniaturized detection in microfluidic chip. Biosens Bioelectron 22:2261–2267
Uchida I, Sekizaki T, Hashimoto K, Terakado N (1985) Association of the encapsulation of Bacillus anthracis with a 60 Megadalton plasmid. J Gen Microbiol 131:363–367
Uchida I, Hashimoto K, Terakado N (1986) Virulence and immunogenicity in experimental animals of Bacillus anthracis strains harbouring or lacking 110 MDa and 60 MDa plasmids. J Gen Microbiol 132:557–559
Wen J, Yang X, Wang K, Tan W, Zuo X, Zhang H (2008) Telomerase catalyzed fluorescent probes for sensitive protein profiling based on one-dimensional microfluidic beads array. Biosens Bioelectron 23:1788–1792
Zaytseva NV, Goral VN, Montagna RA, Baeumner AJ (2005) Development of a microfluidic biosensor module for pathogen detection. Lab Chip 5:805–811
Zhang H, Yang X, Wang K, Tan W, Zhou L, Zuo X, Wen J, Chen Y (2007) Detection of single-base mutations using 1-D microfluidic beads array. Electrophoresis 28:4668–4678
Zhang H, Yang X, Wang K, Tan W, Li H, Zuo X, Wen J (2008) On-chip oligonucleotide ligation assay using one-dimensional microfluidic beads array for the detection of low-abundant DNA point mutations. Biosens Bioelectron 23:945–951
Acknowledgments
This work was supported in part by Canada’s Chemical, Biological, Radiological, and Nuclear Research and Technology Initiative (CRTI) under the program Portable Biological Agent Detection System (06-0187TD). We would like to thank our colleagues and collaborators for useful discussions and technical assistance: Kien-Mun Lau, Maxence Mounier, François Normandin, Boris Le Drogoff, Jean-Guy Allard, Emmanuel Roy, and Hélène Roberge from CNRC Boucherville; Arnold Kell, Chantal Paquet, and Benoît Simard from the Steacie Institute for Molecular Sciences (CNRC, Ottawa, ON); Thompson Tang, William Lee, and Doug Bader from Defense Research and Development Canada (Suffield, Medicine Hat, AB); Ian Summerell from the Royal Canadian Mounted Police (Orleans, ON), Louis Bryden and Michael Mulvey from the Public Health Agency of Canada (Winnipeg, MB); and David Béliveau-Viel, Sébastien Dubus, Even Lemieux, and Prof. Mario Leclerc from Université Laval. We are grateful to Caroline Vachon and Michel M. Dumoulin from CNRC Boucherville for their support.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Jean-François Gravel and Matthias Geissler have contributed equally to this work.
Rights and permissions
About this article
Cite this article
Gravel, JF., Geissler, M., Chapdelaine, S. et al. Portable bead-based fluorescence detection system for multiplex nucleic acid testing: a case study with Bacillus anthracis . Microfluid Nanofluid 16, 1075–1087 (2014). https://doi.org/10.1007/s10404-013-1273-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10404-013-1273-y