
Abstract
This study presents an optical microfluidic platform and method for performing real-time polymerase chain reactions of MDA-MB-231 breast cancer cell DNA within droplet-in-oil micro-reactors. Illumination of the droplets using a low-power (20–40 mW) infrared (IR) laser at 1,460 nm provides a simple approach for droplet manipulation and rapid thermal cycling. The nanoliter droplet volumes allow for extremely fast amplification times, from cell lysis to assay completion in 15 min or less. Droplets containing lysis buffer and subsequently master mix solutions are optically positioned in mineral oil to coalesce with droplets containing live cells on a Petri dish surface for reverse-transcription polymerase chain reactions (RT-PCR). The optical PCR setup is also shown to amplify DNA in droplets containing single or multiple cells and distinguish between methylated and unmethylated BRCA1 promoters in microdroplets containing sample at the single-cell level. Melting curves generated using IR heating indicates a melting temperature of 86 °C for the 255-bp amplicon. The results are consistent with standard PCR and methylation-specific protocols performed in a commercial system. The simplicity of the droplet-in-oil Petri dish platform provides an easy and efficient tool for DNA analysis from live cells, and can be integrated with other microfluidic technologies for complex and large-scale assays.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Since its advent in the mid-1980s, the polymerase chain reaction (PCR) has become a powerful tool in gene analysis and is widely used for gene expression analysis and diagnostics (Erlich et al. 1991). Quantitating transcription levels of specific genes leads to understanding of gene function (Zamorano et al. 1996) and allows for early detection and monitoring of disease progression such as in the case of breast cancer. Circulating tumor cells (CTCs) (Nagrath et al. 2007) may provide a valuable sample type to perform such studies, though single-cell detection levels are required. The reverse-transcription polymerase chain reaction (RT-PCR) is an extremely sensitive method for the detection of low-abundance mRNA in cells (Weis et al. 1992) and permits analysis of samples containing as little as one cell. Methylation-specific PCR (MSP) (Herman et al. 1996) can be another useful method for monitoring disease states due to differential methylation of specific gene promoters in normal and cancer cells. DNA methylation occurs mainly at CpG dinucleotides and involves the enzymatic addition of a methyl group to the cytosine residue without changing the primary DNA sequences (Bird 1980). These modifications at regulatory regions often correspond with changes in the transcriptional state of a gene. Hypermethylation represses transcription, while hypomethylation can lead to increased transcription levels. Investigating such modifications and changes that alter gene expression provides epigenetic information on disease state.
Due to the increasing need of high-throughput cell-based analyses, lab-on-a-chip technologies have been applied to perform PCR on specialized microchips (Northrup et al. 1998; Liu et al. 2004; Roper et al. 2005; Marcus et al. 2006; Lee et al. 2010; Sundberg et al. 2010). Droplet-based microfluidics (Teh et al. 2008) enables the construction of scalable systems that allow the handling of numerous samples or reagents in reconfigurable reactors for biochemical analyses (Shi et al. 2008; Morimoto et al. 2009). High-throughput, cell-based assays in picoliter to nanoliter droplets using microfluidic technologies to rapidly encapsulate biomolecules and cells have recently been reported (Tan et al. 2006). Separated by immiscible oil, the compartmentalization offered in microdroplets allows for hundreds or thousands of emulsion PCR assays to be run concurrently (Nakano et al. 2003; Dorfman et al. 2005; Beer et al. 2007; Ohashi et al. 2007; Kumaresan et al. 2008; Mazutis et al. 2009; Schaerli et al. 2009; Tewhey et al. 2009; Wang and Burns 2009; Zhang et al. 2009; Hatch et al. 2011). The oil also minimizes the risk for cross-contamination of samples. The use of microdroplets in multi-step assays requires simple and efficient actuation methods for transporting, merging, and processing biological samples.
Optical forces as microactuators offer significant benefits in terms of ease in microdroplet discrimination and reconfigurability, and presents unique opportunities for the analysis and control of biological systems such as DNA computation (Nishimura et al. 2012). The full integration of optofluidic techniques for biochemical analysis and on-chip processing is still in its infancy (Hunt and Wilkinson 2008). The purpose of this work is to demonstrate the capability of an optical (laser-based) PCR platform in a clinically relevant study, in particular, mRNA analysis from estrogen receptor negative (ER–) MDA-MB-231 breast cancer cells and determine cancer-specific methylation at the single-cell level in nanoliter volumes.
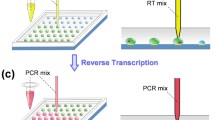
The fundamental scheme of optical manipulation and PCR is illustrated in Fig. 1. The reduced instrument complexity of optical microfluidics (Kotz et al. 2004) allows for the handling of droplets and preceding reactions without external pumps and valves, or complex devices. Optical microdroplet manipulation enables live cell lysis, reverse transcription, and rapid RT-PCR or methylation PCR analysis. Water droplets in oil can be manipulated by thermal Marangoni forces or by convection. For thermal Marangoni motion, nonuniform heating produces a surface tension gradient, and the surface tension gradient induces a net force on the droplet that is required to start motion (Kotz et al. 2004, 2005). For convective motion, laser absorption in mineral oil generates flows in the oil used to manipulate the droplets (Basu and Gianchandani 2008; Dixit et al. 2010; Hu and Ohta 2011; Dixit et al. 2012). PCR can be performed using infrared (IR) light from a lamp, although forced air is typically used to speed the cooling since the entire sample and container are heated (Oda et al. 1998; Hühmer and Landers 2000; Yu et al. 2012). Using an IR laser for PCR is faster still and no active cooling is required when using small droplets. The laser is absorbed by water only, and not in the sample environment (e.g., oil surrounding water droplets, Terazono et al. 2008; Kim et al. 2009a). Notable improvements include controlling droplet shape to minimize the required laser power (Kim et al. 2009a) and modulating the laser power to avoid use of any supporting heater (Kim et al. 2009b). With only an IR laser for thermal cycling, the time for PCR amplification is greatly reduced to 15 min or less.

Conceptual diagram of optical microfluidics. After droplet production, optofluidic techniques are used to combine reagent droplets with the primary cell droplet underneath an oil phase and a defocused IR laser beam is cycled for heating and performing PCR. A measure of fluorescence after each cycle determines amplification level
2 Experimental procedure
2.1 Reagents
Mineral oil, phosphate-buffered saline (PBS), and 0.4 % trypan blue solution were purchased from Sigma (St. Louis, MO). A synthetic lipid, 1,2-diphytanoyl-sn-glycero-3-phosphocholine (DPhPC) was purchased from Avanti Polar Lipids (Alabaster, AL) and was received dissolved in chloroform. Lipid aqueous solutions were prepared by evaporating chloroform from a calculated volume of the stock solution. Hydrated lipid solutions were extruded through a 0.1-μm polycarbonate film in a mini extruder (Avanti Polar Lipids, Alabaster, AL) to yield a 1 mg mL−1 lipid vesicle solution in PBS.
2.2 Cell preparation
The MDA-MB-231 (ER–) cell line is obtained in-house from SRI’s Biosciences Division. Cells are grown in phenol red-free modified Eagle’s medium, supplemented with 10 % fetal bovine serum (Sigma, St. Louis, MO) at 37 °C under 5 % CO2. Prior to lysis, the cells are washed twice with PBS and their concentration is adjusted to 500 cells μL−1. Cell plasma membrane integrity is tracked with the live/dead stain trypan blue, a recognized parameter of cell viability (Life Technologies Corporation, Carlsbad, CA). To determine viability in droplets, 0.1 mL of 0.4 % trypan blue stock solution is added to 0.5 mL of cell suspension. After droplet arrays are printed, the number of blue staining (non-viable) cells is counted at 5 min intervals.
2.3 RT-PCR conditions
RT-PCR is performed for gene expression of 18S rRNA. Cell lysis mixtures consist of 22 μL cell lysis buffer, 2.0 μL DNase I and 1.0 μL RNase inhibitor from a Cells-to-cDNA II kit (Ambion, Austin, TX). PCR master mixtures consist of 10.0 μL PerfeCTa SYBR Green FastMix (Quanta BioSciences, Gaithersburg, MD), and 2.5 μL 10× RT buffer, 1.0 μL RNase inhibitor, 1.0 μL M-MLV reverse transcriptase, 2.0 μL RNA primer, and 4.0 μL RT-PCR grade water (Ambion, Austin, TX). Custom primer sets that target the 1870-base sequence of the human 18S rRNA gene are designed using the OligoPerfect Designer (Life Technologies Corporation, Carlsbad, CA). The size of the PCR amplicon is 133 bp.
2.4 Methylation conditions
Methylation studies are performed on the BRCA1 gene. A DNA methylation enzyme kit containing digestion buffer and restriction enzymes (MEA-03, SABiosciences) is used to prepare genomic DNA samples for predicting methylated CpG islands by real-time PCR. A chemical lysis solution (Cells-to-cDNA II kit, Ambion) is used to create MDA-MB-231 cell lysates. Human male Jurkat (human acute T cell leukemia) genomic DNA (N4002S, New England Biolabs) that is enzymatically methylated with CpG Methylase is used as a positive control for CpG dinucleotide methylation. The concentration is adjusted to that of the cell lysates. No bisulfite modification is necessary for this method.
A 6-h digestion step is performed at 37 °C in a heating block. After incubation, reactions are stopped by heat-inactivating the enzymes at 65 °C for 20 min. DNA samples are used immediately or stored at –20 °C for future experiments. Human BRCA1 qPCR primers targeting the CpG island location on chromosome 17 (38530718–38531083) are used in the DNA methylation study (MePH28472-1B, SABiosciences). The size of the PCR amplicon is 255 bp. The one-step target gene amplification master mixture contains 2 μL cell digest, 12 μL of SYBR Green/ROX PCR Master Mix (PerfeCTa SYBR Green FastMix, Quanta), and 6.0 μL RT-PCR grade water (Ambion, Austin, TX).
2.5 Instrumentation
2.5.1 Commercial PCR setup
The ABI Prism 7500 (Applied Biosystems, Foster City, CA) contains a built-in thermal cycler with 96-well positions, and is able to detect fluorescence between 500 and 660 nm. Aliquots of 20 μL (18 μL master mix + 2 μL cell digest) are added to a thin-walled MicroAmp optical 96-well reaction plate (Applied Biosystems, Foster City, CA). The DNA digests are subjected to 40 PCR amplification cycles. All samples are amplified in duplicates. As a negative control, a PCR reaction is carried out in the absence of a template.
Thermal cycling conditions for RT-PCR using 18S RNA primers comprise 15 min at 45 °C for reverse transcription, 3 min at 95 °C followed by 40 cycles of 20 s at 95 °C and 40 s at 60 °C. Thermal cycling conditions using BRCA1 primers comprise 2 min at 50 °C, 10 min at 95 °C followed by 40 cycles of 15 s at 97 °C and 1 min at 72 °C. Fluorescence generation due to SYBR Green chemistry is measured sequentially for each sample after each cycle. PCR reactions typically take over 1.5 h to complete due to slow ramp times. Absolute quantification studies are made from collected data with the Applied Biosystems 7500 System SDS software 1.3.
2.5.2 Optical PCR setup
An inverted fluorescence microscope (TE2000, Nikon Instruments) containing a dual filter-turret and fitted with a 10× Nikon objective is used for imaging and all steps of PCR. Figure 2 illustrates the system design. The top filter cube is used for fluorescence excitation and emission collection and the bottom filter cube delivers an IR beam from a diode laser (FOL 1402PLY-617-1457, Furukawa Electric) of wavelength 1.46 μm for heating the droplet. A current source (LDX-3620, ILX Lightwave) under computer control varies the laser power for thermal cycling. Band pass filters are used for fluorescence detection with a 480/40-nm filter for excitation and a 520/40-nm filter for emission. All dichroics and filters are purchased from Chroma (Bellows Falls, VT). A 4-f two lens configuration based on a Thorlabs cage system is utilized to couple the laser to the microscope. Defocusing the laser beam at the droplets is achieved by translating one lens in order to achieve a ~300-μm beam size and obtain a relatively uniform temperature distribution. The laser beam position and size are monitored using a transparent tape coated with a thin layer of erbium upconverting phosphor powder. Bright-field and fluorescence images are acquired with a cooled CCD camera (Retiga EXi, QImaging). All experiments are conducted in clear, 60 mm polystyrene Petri dishes (Nunc, Thermo Fisher Scientific) at ambient temperature (23 °C).

Instrument schematic for optical PCR with fluorescence detection. The system is controlled with a LabVIEW computer program
2.6 Microdroplet PCR
A 10-μL micropipette tip is used to produce droplets with diameters as small as 200 μm at pitches of 1 mm or less between droplets. No treatment is necessary to prevent adsorption of DNA to the polystyrene surface. While printing, the droplet array is permanently protected from evaporation by the immediate addition of a sterile and filtered mineral oil. Mineral oil is used because it has a lower density (0.82–0.88 g mL−1) than water, has extremely low water solubility, and prevents droplet evaporation.
Droplet temperatures are calibrated based on fluorescence measurements of SYBR Green as well as amplification efficiency. Fluorescence quenching is used to calibrate the laser power required to heat the droplet up to the annealing/extension temperature and melting temperature (Kim et al. 2009a). By variation of the laser power between droplets, the temperature range based on PCR results is determined. The laser power is therefore used as an indicator of the droplet temperature. RT-PCR comprises 3 min at 42 °C (125 mA/15 mW) for reverse transcription followed by a 12-min laser-based thermal cycling step controlled by LabVIEW. Thermal cycling consists of 20 s at 95 °C (400 mA/47 mW) for denaturation/enzyme activation followed by 50 cycles of 3 s at 95 °C (400 mA/47 mW) and 12 s at 55 °C (185 mA/20.5 mW). Methylation-specific PCR comprises 10 s at 95 °C (400 mA/47 mW) followed by 50 cycles of 3 s at 97 °C (420 mA/48 mW) and 12 s at 72 °C (300 mA/34 mW). Droplets are imaged during the final second of the annealing phase of each cycle for real-time SYBR Green fluorescence detection, controlled in LabVIEW. The average pixel intensity for each droplet is tracked through 50 cycles of PCR. Fluorescence images are acquired at a 400-ms acquisition time to avoid observable fluorophore photobleaching effects. The fluorescence illumination source is blocked with an automated shutter between fluorescence measurements.
2.7 Imaging and analysis
Each droplet is identified and sized from a bright-field image. The polydispersity index σ = δ/d avg × 100 % was calculated from the average droplet size d avg and standard deviation δ, determined by measuring droplet sizes from recorded images. The Ct value or threshold cycle (where there is first clearly detectable increase in fluorescence) is obtained from the exponential phase of the fluorescence for each run. When using our optical setup, the threshold fluorescence level is set manually at a level sufficiently above the baseline to prevent threshold crossing by noise and sufficiently low to be within the exponential growth region of the amplification curve. The commercial PCR system automatically sets the threshold fluorescence. For both PCR amplification and melting analysis, ImageJ (Abràmoff et al. 2004) is used to perform an intensity measurement for each droplet image over time. All the raw fluorescence readings of individual PCR amplifications are fitted to the four-parametric sigmoid function using the nonlinear regression function of SigmaPlot 12 (Systat Software, San Jose, CA). The melting temperature, T m, defined as the temperature where the negative value of the derivative of the fluorescence signal gain over temperature is at its maximum, is used to distinguish the sample DNA amplicons from non-specific products through variations in melting temperatures. Melting curve analysis consists of 80 intervals at 500-ms exposure starting at 40 °C (115 mA/10 mW) and ending at 98 °C (434 mA/50 mW).
3 Results and discussion
3.1 Optical RT-PCR
Conditions for cell lysis and RT-PCR using a multi-droplet deposition scheme on a Petri dish were investigated. Laser manipulation of droplets is made possible by thermal Marangoni effects or convection due to heating, as described in “Introduction”. The behavior of a droplet is sensitive to its size, and droplets with different sizes have different velocities under the same laser power. A medium-sized (~300 μm) droplet moves slowly in a circular Petri dish compared with a smaller (~150 μm) droplet. The velocity is also dependent on the viscosity of the external oil. When the laser is turned off, the viscous drag will decelerate the droplet. In the case of mineral oil, within seconds the droplets settle. Droplets containing the cell sample must remain adhered to the bottom Petri dish surface and remain immobile after droplet coalescence and during the thermal cycling stage of PCR. The lysis and PCR reagent droplets should have as small a contact area as possible for manipulation with the laser. This was made possible by the addition of a surfactant or lipid (DPhPC) layer to the lysis and reagent droplets, ensuring that they remain mobile. By positioning the laser at the site of the cell droplet, it is possible to direct the lysis and PCR reagent droplets toward the cell droplet.
A multi-tip deposition was used for droplet array production on Petri dishes. Each analyzed sessile PBS droplet containing live cells (Fig. 3a) was approximately 40 nL in volume (diameter of ~330 μm and σ < 15 %), with a contact angle of approximately 110° under mineral oil on a Nunc polystyrene tissue culture plate. Prior viability measurements from the reactivity of trypan blue dye confirmed that cells remained viable within the droplet microenvironment for at least 30 min. Lipid-coated droplets containing lysis buffer were optically positioned in mineral oil and forced to coalesce with droplets containing live cells on a Petri dish surface. This chemical lysis solution was utilized to completely lyse cells as laser-assisted droplet heating (during the hot start step for polymerase activation) was not sufficient to release the genomic DNA. A PCR reagent droplet was then optically positioned and forced to coalesce with the lysed cell droplet, followed by optical RT-PCR with 18S RNA primers (Fig. 3b) for 50 cycles.

Droplet manipulation on a Petri dish and PCR. a After spotting PBS microdroplets containing single cells (1) dispersed in oil, microdroplets containing lysis buffer (2, 3) and PCR reagents (4, 5) can be moved and mixed using an IR laser. The defocused laser beam is cycled for heating and performing PCR (6) within the microdroplet. b Montage of 50 fluorescence images of a single droplet during a PCR run
Optical RT-PCR amplification is shown from droplets containing 1, 3, and 7 cells versus that of the control (Fig. 4). Since PCR amplification is an exponential reaction, plotting these curves in the standard fashion makes single-cell amplification difficult to detect on the same scale when compared with droplets containing three and seven cells. Amplifications were reproducible with optical RT-PCR, but Ct values were found to differ for each condition, possibly due to variations in droplet size (leads to unequal amounts of reagents, primers, and probes) and the degree of success in cell lysis (important for maintaining the integrity and expected amount of sample DNA from droplet to droplet). The volumes that can be analyzed are 105–106 times smaller than in commercial real-time PCR systems. RT-PCR is inhibited if the concentration of cells exceeds a threshold number. Serial dilutions of cells were made to determine the optimum and maximum cells that can be used in reactions without inhibiting reverse transcription. At cell concentrations above 1,000 cells per microliter of lysis buffer, the PCR signal was clearly diminished, possibly due to lack of complete RNase inhibition or the presence of high amounts of cell debris. In addition, when the volume of lysis buffer was less than 20 % of the total RT-PCR mixture, on-chip RT-PCR was successfully performed. At 30–40 % of the total mixture volume, the product yield from reverse transcription was visibly reduced (data not shown).

Live cells encapsulated in microdroplets and PCR amplification. a Microdroplets containing various cell numbers and b subsequent PCR results
3.2 DNA methylation analysis
The BRCA1 protein plays a major role in DNA double-strand break repair through homologous recombination, and it is well known that females that have BRCA1 mutations, and more specifically BRCA1 methylation, have a significantly elevated lifetime risk of breast cancer (Yang et al. 2001; Thakur et al. 2003). The degree of BRCA1 methylation was experimentally determined for the estrogen receptor negative (ER–) breast cancer cell line MDA-MB-231. Human male Jurkat (human acute T cell leukemia) genomic DNA that was enzymatically methylated was used as a positive control. A multi-tip deposition was used for droplet array production on Petri dishes. The cell concentration was adjusted to ensure that each droplet contained the contents of a single cell.
Often, the DNA template has to be chemically modified with the use of sodium bisulfite to preserve methylation information before PCR amplification. To avoid this time-consuming step, the Methyl-Profiler™ DNA Methylation PCR System (SABiosciences Corporation) was used for detection of CpG island DNA methylation profiles of the BRCA1 gene. This technology is based on the detection of remaining input DNA after cleavage with a methylation-sensitive and/or a methylation-dependent restriction enzyme. Following digestion (of unmethylated and/or methylated DNA), the remaining DNA is quantified by real-time PCR in each individual enzyme reaction using primers that flank the BRCA1 gene region of interest. The relative fractions of hypermethylated, intermediately methylated, and unmethylated DNA are determined by comparing the amount in each digest with a mock (no enzymes added) digest. As shown in Fig. 5, the real-time fluorescence intensity curves for each sample droplet exhibits exponential, linear, and plateau phases. The PCR threshold cycle (Ct) and amount of input DNA have an inversely proportional relationship with the mock digest (Mo) Ct values, single enzyme digest (methylation-sensitive and methylation-dependent, Ms and Md, respectively) Ct values, and double digest (Msd) Ct values using our optical PCR setup. The difference in Ct values between the double and mock digests was greater than five [ΔCt (Msd − Mo) > 5], meaning that more than 97 % of all DNA molecules in the samples from both systems were digested.

Methylation PCR data showing fluorescence intensity versus cycle number for microdroplet sets using the optical PCR system and a commercial instrument for (a) MDA-MB-231 and (b) Jurkat DNA, respectively
For the MDA-MB-231 cell DNA, we observed ΔCt (Md − Mo) < 1.0 and ΔCt (Ms − Mo) > 1.0 (Fig. 5a), while for the Jurkat cell DNA we observed ΔCt (Ms − Mo) < 1.0 and ΔCt (Md − Mo) > 1.0 (Fig. 5b) using the optical PCR setup. These results were consistent with what was observed using a commercial instrument. Table 1 lists the Ct values used for the DNA methylation analysis. The formulas and equations used to calculate the methylation status of BRCA1 can be found in the Appendix of the “Methyl-Profiler™ DNA Methylation PCR Array System” user manual by SABiosciences Corporation. Table 2 displays the relative percentage of hypermethylated (HM), intermediately methylated (IM), and unmethylated (UM) DNA for the target sequence (BRCA1). Each row in Tables 1 and 2 represent a separate data set. The optical PCR analysis represents data from lysates at a single-cell concentration, whereas the commercial system analysis represents data for a population of approximately 500 cells. No BRCA1 methylation was found in the MDA-MB-231 DNA after multiple runs with our optical PCR setup and was confirmed with a commercial instrument. For enzymatically methylated Jurkat DNA, results from both systems confirm >90 % hypermethylation. The lack of BRCA1 promoter region methylation in MDA-MB-231 (and most other breast cancer cell lines) indicates that the region is resistant to non-specific methylation. However, cancer cell lines often acquire de novo methylation (Bianco et al. 2000). Recent studies (Wei et al. 2005) indicate that sporadic breast tumors are methylated at the BRCA1 promoter region, leading to the lack of the protein.
We ran multiple dissociation experiments following the real-time PCR for each droplet with SYBR Green-based detection to ensure that the correct amplicon was detected. As an intercalating dye, SYBR Green will detect any double-stranded DNA including primer dimers and contaminating DNA. A plot of the melting curves when using BRCA1 is shown in Fig. 6a. Dissociation (melting temperature) curves are shown for both MDA-MB-231 DNA and Jurkat DNA (four curves for each sample type) from different droplets that had just previously undergone methylation PCR under different conditions as shown in Fig. 5. For the analysis, the droplet temperature is incrementally raised by a fraction of a degree and the differential change in fluorescence is measured (Fig. 6b). At the melting point, the DNA strands will separate and the fluorescence rapidly decreases. The melting temperatures of primers start at the point of inflection of the melting curve profile. We determined that this point occurs at approximately 86 °C for BRCA1. Droplet positions were stable during amplification and melting curve analysis. Automation of all steps from enzyme activation through final analysis makes optical PCR simple to perform.

Dissociation data. a Melting curves showing fluorescence intensity versus temperature for droplet sets using the optical PCR setup for both MDA-MB-231 and Jurkat DNA, respectively. b Montage of fluorescence images of a single droplet during an 80-interval melting analysis
3.3 Amplification issues
For testing cross-contamination between droplets, solutions were made that do not contain DNA template or primer. The lack of amplification in control droplets confirms that there is no observable contamination between different droplets. We find that the amplification of methylated and unmethylated DNA is comparable to that obtained in a conventional thermal cycler for all positive starting concentrations at or above the single-cell level. However, the efficiency can vary between droplets at starting concentrations below the single-cell level due to lack of DNA copies. This is largely due to a Poisson distribution, which can affect the amount of template in each sessile droplet (Kiss et al. 2008). Microfluidic methods for achieving sub-Poisson statistics of cells in droplets have been developed (Edd et al. 2008; Abate et al. 2009). We also initially ran into problems with temperature nonuniformity across the droplets that impacts amplification efficiency. A 4-f lens configuration was then utilized to defocus the laser beam to achieve a more uniform temperature distribution.
4 Conclusion
Each 50-cycle PCR or RT-PCR experiment in droplets can be performed in less than 15 min for a range of amplicon sizes using the optical microfluidic setup, compared with approximately 1.5 h for a 40-cycle experiment on the commercial system. The simplicity of the droplet-in-oil optical microfluidic PCR platform can therefore provide an easy and efficient tool for DNA analysis of breast tumor cells in large-scale arrays. The low-power (20–40 mW) system is a candidate for further miniaturization and automation. We plan to develop an integrated system for cell encapsulation in microdroplets, lysis, enzyme digestion, and real-time PCR amplification using optical droplet manipulation.
References
Abate AR, Chen CH, Agresti JJ, Weitz DA (2009) Beating Poisson encapsulation statistics using close-packed ordering. Lab Chip 9:2628–2631
Abràmoff MD, Magalhães PJ, Ram SJ (2004) Image processing with ImageJ. Biophotonics Int 11:36–42
Basu AS, Gianchandani YB (2008) Virtual microfluidic traps, filters, channels and pumps using Marangoni flows. J Micromech Microeng 18:115031
Beer NR, Hindson BJ, Wheeler EK, Hall SB, Rose KA, Kennedy IM, Colston BW (2007) On-chip, real-time, single-copy polymerase chain reaction in picoliter droplets. Anal Chem 79:8471–8475
Bianco T, Chenevix-Trench G, Walsh DC, Cooper JE, Dobrovic A (2000) Tumour-specific distribution of BRCA1 promoter region methylation supports a pathogenetic role in breast and ovarian cancer. Carcinogenesis 21:147–151
Bird AP (1980) DNA methylation and the frequency of CpG in animal DNA. Nucleic Acids Res 8:1499–1504
Dixit SS, Kim H, Vasilyev A, Eid A, Faris GW (2010) Light-driven formation and rupture of droplet bilayers. Langmuir 26:6193–6200
Dixit SS, Pincus A, Guo B, Faris GW (2012) Droplet shape analysis and permeability studies in droplet lipid bilayers. Langmuir 28:7442–7451
Dorfman KD, Chabert M, Codarbox J-H, Rousseau G, de Cremoux P, Viovy J-L (2005) Contamination-free continuous flow microfluidic polymerase chain reaction for quantitative and clinical applications. Anal Chem 77:3700–3704
Edd JF, Di Carlo D, Humphry KJ, Koster S, Irimia D, Weitz DA, Toner M (2008) Controlled encapsulation of single-cells into monodisperse picolitre drops. Lab Chip 8:1262–1264
Erlich HA, Gelfand D, Sninsky JJ (1991) Recent advances in the polymerase chain reaction. Science 252:1643–1651
Hatch AC, Fisher JS, Pentoney SL, Yang DL, Lee AP (2011) Tunable 3D droplet self-assembly for ultra-high-density digital micro-reactor arrays. Lab Chip 11:2509–2517
Herman JG, Graff JR, Myöhänen S, Nelkin BD, Baylin SB (1996) Methylation-specific PCR: a novel PCR assay for methylation status of CpG islands. Proc Natl Acad Sci USA 93:9821–9826
Hu W, Ohta A (2011) Aqueous droplet manipulation by optically induced Marangoni circulation. Microfluid Nanofluid 11:307–316
Hühmer AFR, Landers JP (2000) Noncontact infrared-mediated thermocycling for effective polymerase chain reaction amplification of DNA in nanoliter volumes. Anal Chem 72:5507–5512
Hunt HC, Wilkinson JS (2008) Optofluidic integration for microanalysis. Microfluid Nanofluid 4:53–79
Kim H, Dixit S, Green CJ, Faris GW (2009a) Nanodroplet real-time PCR system with laser assisted heating. Opt Express 17:218–227
Kim H, Vishniakou S, Faris GW (2009b) Petri dish PCR: laser-heated reactions in nanoliter droplet arrays. Lab Chip 9:1230–1235
Kiss MM, Ortoleva-Donnelly L, Beer NR, Warner J, Bailey CG, Colston BW, Rothberg JM, Link DR, Leamon JH (2008) High-throughput quantitative polymerase chain reaction in picoliter droplets. Anal Chem 80:8975–8981
Kotz KT, Noble KA, Faris GW (2004) Optical microfluidics. Appl Phys Lett 85:2658–2660
Kotz KT, Gu Y, Faris GW (2005) Optically addressed droplet-based protein assay. J Am Chem Soc 127:5736–5737
Kumaresan P, Yang CJ, Cronier SA, Blazej RG, Mathies RA (2008) High-throughput single copy DNA amplification and cell analysis in engineered nanoliter droplets. Anal Chem 80:3522–3529
Lee D, Chen P-J, Lee G-B (2010) The evolution of real-time PCR machines to real-time PCR chips. Biosens Bioelectron 25:1820–1824
Liu RH, Yang J, Lenigk R, Bonanno J, Grodzinski P (2004) Self-contained, fully integrated biochip for sample preparation, polymerase chain reaction amplification, and DNA microarray detection. Anal Chem 76:1824–1831
Marcus JS, Anderson WF, Quake SR (2006) Microfluidic single-cell mRNA isolation and analysis. Anal Chem 78:3084–3089
Mazutis L, Araghi AF, Miller OJ, Baret J-C, Frenz L, Janoshazi A, Taly V, Miller BJ, Hutchison JB, Link D, Griffiths AD, Ryckelynck M (2009) Droplet-based microfluidic systems for high-throughput single DNA molecule isothermal amplification and analysis. Anal Chem 81:4813–4821
Morimoto Y, Tan W-H, Tsuda Y, Takeuchi S (2009) Monodisperse semi-permeable microcapsules for continuous observation of cells. Lab Chip 9:2217–2223
Nagrath S, Sequist LV, Maheswaran S, Bell DW, Irimia D, Ulkus L, Smith MR, Kwak EL, Digumarthy S, Muzikansky A, Ryan P, Balis UJ, Tompkins RG, Haber DA, Toner M (2007) Isolation of rare circulating tumour cells in cancer patients by microchip technology. Nature 450:1235–1239
Nakano M, Komatsu J, Matsuura S-I, Takashima K, Katsura S, Mizuno A (2003) Single-molecule PCR using water-in-oil emulsion. J Biotechnol 102:117–124
Nishimura T, Ogura Y, Tanida J (2012) Optofluidic DNA computation based on optically manipulated microdroplets. Microfluid Nanofluid. doi:10.1007/s10404-012-0934-6
Northrup MA, Benett B, Hadley D, Landre P, Lehew S, Richards J, Stratton P (1998) A miniature analytical instrument for nucleic acids based on micromachined silicon reaction chambers. Anal Chem 70:918–922
Oda RP, Strausbauch MA, Huhmer AFR, Borson N, Jurrens SR, Craighead J, Wettstein PJ, Eckloff B, Kline B, Landers JP (1998) Infrared-mediated thermocycling for ultrafast polymerase chain reaction amplification of DNA. Anal Chem 70:4361–4368
Ohashi T, Kuyama H, Hanafusa N, Togawa Y (2007) A simple device using magnetic transportation for droplet-based PCR. Biomed Microdevices 9:695–702
Roper MG, Easley CJ, Landers JP (2005) Advances in polymerase chain reaction on microfluidic chips. Anal Chem 77:3887–3893
Schaerli Y, Wootton RC, Robinson T, Stein V, Dunsby C, Neil MAA, French PMW, Demello AJ, Abell C, Hollfelder F (2009) Continuous-flow polymerase chain reaction of single-copy DNA in microfluidic microdroplets. Anal Chem 81:302–306
Shi W, Qin J, Ye N, Lin B (2008) Droplet-based microfluidic system for individual Caenorhabditis elegans assay. Lab Chip 8:1432–1435
Sundberg SO, Wittwer CT, Gao C, Gale BK (2010) Spinning disk platform for microfluidic digital polymerase chain reaction. Anal Chem 82:1546–1550
Tan Y-C, Hettiarachchi K, Siu M, Pan Y-R, Lee AP (2006) Controlled microfluidic encapsulation of cells, proteins, and microbeads in lipid vesicles. J Am Chem Soc 128:5656–5658
Teh S-Y, Lin R, Hung L-H, Lee AP (2008) Droplet microfluidics. Lab Chip 8:198–220
Terazono H, Hattori A, Takei H, Takeda K, Yasuda K (2008) Development of 1,480 nm photothermal high-speed real-time polymerase chain reaction system for rapid nucleotide recognition. Jpn J Appl Phys 47:5212–5216
Tewhey R, Warner JB, Nakano M, Libby B, Medkova M, David PH, Kotsopoulos SK, Samuels ML, Hutchison JB, Larson JW, Topol EJ, Weiner MP, Harismendy O, Olson J, Link DR, Frazer KA (2009) Microdroplet-based PCR enrichment for large-scale targeted sequencing. Nat Biotechnol 27:1025–1031
Thakur S, Nakamura T, Calin G, Russo A, Tamburrino JF, Shimizu M, Baldassarre G, Battista S, Fusco A, Wassell RP, Dubois G, Alder H, Croce CM (2003) Regulation of BRCA1 transcription by specific single-stranded DNA binding factors. Mol Cell Biol 23:3774–3787
Wang F, Burns MA (2009) Performance of nanoliter-sized droplet-based microfluidic PCR. Biomed Microdev 11:1071–1080
Wei M, Grushko TA, Dignam J, Hagos F, Nanda R, Sveen L, Xu J, Fackenthal J, Tretiakova M, Das S, Olopade OI (2005) BRCA1 promoter methylation in sporadic breast cancer is associated with reduced BRCA1 copy number and chromosome 17 aneusomy. Cancer Res 65:10692–10699
Weis JH, Tan SS, Martin BK, Wittwer CT (1992) Detection of rare mRNAs via quantitative RT-PCR. Trends Genet 8:263–264
Yang X, Yan L, Davidson NE (2001) DNA methylation in breast cancer. Endocr-Relat Cancer 8:115–127
Yu Y, Li B, Baker CA, Zhang X, Roper MG (2012) Quantitative polymerase chain reaction using infrared heating on a microfluidic chip. Anal Chem 84:2825–2829
Zamorano PL, Mahesh VB, Brann DW (1996) Quantitative RT-PCR for neuroendocrine studies: A mini review. Neuroendocrinology 63:397–407
Zhang Y, Bailey V, Puleo CM, Easwaran H, Griffiths E, Herman JG, Baylin SB, Wang T-H (2009) DNA methylation analysis on a droplet-in-oil PCR array. Lab Chip 9:1059–1064
Acknowledgments
We thank Nahid Waleh for technical assistance, and acknowledge the thoughtful discussions with Keith Laderoute, Christopher Green, and Sanhita Dixit. This work was supported by the DOD Breast Cancer Research Program, Grant Number W81XWH-09-1-0298. The US Army Medical Research Acquisition Activity, 820 Chandler Street, Fort Detrick MD 21702-5014 is the awarding and administering acquisition office. The content of this paper does not necessarily reflect the position or the policy of the government, and no official endorsement should be inferred.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Hettiarachchi, K., Kim, H. & Faris, G.W. Optical manipulation and control of real-time PCR in cell encapsulating microdroplets by IR laser. Microfluid Nanofluid 13, 967–975 (2012). https://doi.org/10.1007/s10404-012-1016-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10404-012-1016-5