
Abstract
We report an active micromixer utilizing vortex generation due to non-equilibrium electrokinetics near micro/nanochannel interfaces. Its design is relatively simple, consisting of a U-shaped microchannel and a set of nanochannels. We fabricated the micromixer just using a two-step reactive ion etching process. We observed strong vortex generation in fluorescent microscopy experiments. The mixing performance was evident in a combined pressure-driven and electroosmotic flows, compared with the case with a pure pressure-driven flow. We characterized the micromixer for several conditions: different applied voltages, ion concentrations, flow rates, and nanochannel widths. The experimental results show that the mixing performance is better with a higher applied voltage, a lower ion concentration, and a wider nanochannel width. We quantified the mixing characteristics in terms of mixing time. The lowest mixing time was 2 milliseconds with the voltage of 230 V and potassium chloride solutions of 0.1 mM. We expect that the micromixer is beneficial in several applications requiring rapid mixing.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Mixing different fluids at micro/nanoscale is one of the challenges in microfluidic technology (Di Carlo 2009). Many researchers have extensively studied and developed various micromixers since the past decade. Micromixers have been applied to, e.g., lab-on-a-chip devices, drug delivery, biochemical detection systems, nucleic acid sequencing, and microchemical synthesis (Haeberle and Zengerle 2007; Mansur et al. 2008; Min et al. 2004; Schulte et al. 2002; Yi et al. 2006). A variety of micromixers has been proposed and demonstrated in response to diverse needs, e.g., reduced analysis time, improved mixing efficiency, and controlled chemical process (Miller and Wheeler 2008; Puleo and Wang 2009). Rapid micromixing is beneficial in some of biochemical fields such as protein folding. Reagents should be rapidly mixed in such applications before a significant reaction process begins (Gambin et al. 2010; Neils et al. 2004; Ridgeway et al. 2009; Roder et al. 2004; Shcherbakova and Brenowitz 2008).
Micromixers can be classified into passive and active mixers according to the existence of external energy input (Nguyen and Wu 2005; Chen and Cho 2008). Passive mixers do not require external energy input and they generate mixing with the help of chaotic advection, diffusion, etc. This type of mixer typically relies on complex flow patterns, which define unique structures of different passive mixers. Various passive micromixers have been reported: branched-microchannels (Erbacher et al. 1999), staggered herringbone mixers (Stroock et al. 2002; Villermaux et al. 2008), zigzag-shaped channels (Chen and Yang 2007; Egawa et al. 2009), three-dimensional nanopore structures (Park et al. 2009), and split-and-recombine configurations (Hardt et al. 2005, 2006; Tofteberg et al. 2010). Active mixers, on the other hand, operate with external energy input and thus mixing is achieved with various external disturbances. The advantages include controlled mixing conditions, Péclet number-independent operation (sometimes with very low Péclet number), and improved mixing efficiency (Hessel et al. 2005). This type of mixer, however, requires extra fabrication steps and additional experimental setup often (Hsiung et al. 2007; Niu et al. 2006). Active micromixers can be categorized according to external energy inputs: acoustic disturbance (Frommelt et al. 2008; Ahmed et al. 2009; Yaralioglu et al. 2004), dielectrophoretic disturbance (Gadish and Voldman 2006; Lee and Voldman 2007), asymmetric post arrays (Hsiung et al. 2007; Wang et al. 2007), electrohydrodynamic instability (El Moctar et al. 2003), electroosmotic disturbance (Sasaki et al. 2006, 2010), static magnetic field (Goet et al. 2009), magnetic immobilization of magnetic beads (Hirschberg et al. 2009; Lund-Olesen et al. 2008), and electrokinetically driven mixing (Lee et al. 2004; Yan et al. 2009; Wu and Li 2008).
We report an active micromixer which utilizes vortex generation due to non-equilibrium electrokinetics near micro/nanochannel interfaces. Recently, there have been extensive studies on so-called concentration polarization (CP) and non-equilibrium electrokinetic phenomena near micro/nanochannel interfaces (Dukhin 1993; Rubinstein et al. 2008; Zaltzman and Rubinstein 2007). The application of electric fields across nanochannels creates ion enrichment at one end and ion depletion at the other end. This effect, called concentration polarization, occurs when ionic currents are blocked through nanochannels (Kim et al. 2009; Mani et al. 2009). In nanochannels, there is an overlap of charge-shielding layers, which is called electric double layers (EDLs) (Kim et al. 2007, 2010). CP affects ion perm- selectivity, conductivity, and electric field profiles around micro/nanochannel interfaces as it changes ion transport (Mani et al. 2009; Choi and Kim 2009; Plecis et al. 2008). A further increased electric field results in the breakup of a CP structure, generating vortices near micro/nanochannel interfaces (Yossifon and Chang 2008). The strong vortices are the result of electrokinetics at its non-equilibrium state.
Pu et al. (2004) visualized CP phenomena around nanochannel areas and presented a qualitative model to explain their observation. Kim et al. (2007) studied these phenomena with their precisely fabricated micro/nanochannel devices and visualized detailed vortex structures in a high electric field regime. Park et al. (2006) reported that a recirculation effect generates vortices near micro/nanochannel interfaces in their computation study. These vortices are due to an internal pressure gradient: it is formed by non-uniform electro-osmotic flow fields with curved ionic currents through the cross-sectional area-varying channel. Rubinstein and his co-workers found that electrokinetic flow instability can be a mechanism for the formation of vortices (Rubinshtein et al. 2002; Zaltzman and Rubinstein 2007). Their theoretical study showed that vortices explain an overlimiting current through a nanoporous membrane.
Non-equilibrium electrokinetics (vortex generation at high electric field) was applied as a microfluidic mixer. Kim et al. (2008) proposed a practical design which allows efficient vortex generation at micro/nanochannel interfaces. This design is simple in a small space with no moving part, which is beneficial for integration with other microfluidic systems. A relatively easy device fabrication would be also advantageous for lab-on-a-chip system application. Kim et al. (2008) performed mixing experiments for different electrolyte concentrations and applied voltages. Their demonstration showed the potential application of this type of mixer as an efficient micromixing technique. However, their experiments were only with relatively low flow rate ranges because the driving force was only an electroosmotic flow (EOF). They did not report the mixing time of the mixer, which is one of the most important parameters for micromixers.
In this paper, we demonstrated excellent performance of a non-equilibrium electrokinetic mixer with fast pressure-driven flows (PDF). We especially quantified mixing time of our mixer in different operating conditions, e.g., concentrations and electric fields. We also varied flow rates and nanochannel types. The fastest mixing time was measured in the order of a millisecond for many cases.
2 Experimental
The non-equilibrium electrokinetic microfluidic mixer consists of a U-shaped microchannel and a set of nanochannels across the two sections of this microchannel, as shown in Fig. 1. The microchannel has two inlet streams, each of which contains fluids to be mixed. The purpose of the U-shaped microchannel is to serve as a flow passage and the inlet/outlet of nanochannels rather than to serve as a mixing channel. Mixing would occur in fluid flows downstream along the microchannel because of spontaneous molecular diffusion. However, the length of the microchannel with only one turn is insufficient for mixing, as suggested by the work by Chen and Yang. They characterized mixing performance of zigzag microchannels and they found that their device needs many turns for perfect mixing to achieve enough diffusion length.

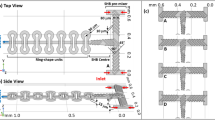
Schematic of a microfluidic mixer (left) and microscope images of four types of nanochannel patterns (right). The mixer is composed of a U-shaped microchannel and nanochannels across the two microchannel sections. The microchannel has two inlets (for two fluids to be mixed) and one outlet
An advantage of the non-equilibrium electrokinetic mixer is its potential easy integration with other microfluidic components. The U-shaped microchannel can be fabricated in the same process together with other components and the only additional fabrication would be nanochannel fabrication. The device is just two-layered with all the features on a silicon wafer. A flat glass wafer is bonded to form a closed channel and to give optical access. This two-layered design allows the avoidance of sometimes troublesome multi-layer fabrication, which is required for a certain type of passive mixer (Nguyen and Wu 2005).
The microchannel has a depth of 13 μm, a width of 100 μm, and an overall length of roughly 1.5 cm. The distance between two microchannel sections, across which a set of nanochannels are located, is 50 μm. This distance coincides with the nanochannel length. The nanochannels are 55 nm in depth with varying widths. We prepared four different types of nanochannels: (1) 10 sets and (2) 100 sets of 10-μm-wide nanochannels with 10-μm-wide spacings and (3) 3 sets and (4) 17 sets of alternating 10-μm- and 50-μm-wide nanochannels with 10-μm-wide spacings (see Fig. 1). Figure 2 shows a scanning electron microscope (SEM) image of the cross section of a nanochannel. We here observe that the height of a nanochannel is uniform albeit with some roughness.

Scanning electron microscope (SEM) image of a nanochannel cross-section
We made the non-equilibrium electrokinetic mixer with silicon-based nanofabrication techniques. We fabricated all the micro- and nanochannels as well as fluidic access holes on a silicon wafer. We then bonded a Pyrex glass wafer to the silicon wafer to finish closed micro/nanofluidic channels. We needed three photomasks to pattern the microchannel, the nanochannels, and the access holes. The silicon wafers were 4-inch-long, 550-μm-thick, arsenic-doped N-type, prime-grade, <1–0–0> wafers. The glass wafers were 4-inch-long, 550-μm thick Pyrex wafers. Indeed, any glass wafer allowing anodic bonding with a silicon wafer is acceptable for our mixer, since there is no feature on a glass wafer.
We performed photolithography and etching in the order of the nanochannels, the microchannel, and the access holes. The photolithography process was typical: spin coating of photoresist (PR), mask alignment, development, and soft/hard bake processes. The etching for all the features was reactive ion etching (RIE) with an ICP-DRIE system (Plasmatherm SLR-770). We had a slow etching rate for nanochannels to obtain smooth nanochannel surfaces. The major parameters in the nanochannel recipe were: 20 sccm SF6, 40 sccm O2, 40 sccm Ar, and a RF power of 500 W. We used the Bosch deep reactive ion etching (DRIE) for microchannels and through-holes for fluidic access. The silicon wafer has only native oxides after all the etching processes and we needed oxidation for electrical insulation. Before thermal oxidation, we took a careful cleaning step using the RCA-1 solution. A roughly 350-nm-thick oxide layer was thermally grown at 1,100°C for about 14 h in a wet oxygen-fed tube furnace. The last step in the nanofabrication was anodic bonding of the patterned Si wafer with the glass wafer (AST Tbon-100 bonder). We executed low-temperature anodic bonding with 354°C, 800 V, and 2,000 N to avoid nanochannel collapse. Mao and Han extensively studied the temperatures required to maintain nanochannels for different widths and depths of nanochannels while avoiding nanochannel collapse (Mao and Han 2005).
We used potassium chloride (KCl) solutions of 0.1–50 mM as a working fluid. The two inlet streams contain KCl solutions of the identical concentration. A minute amount (~0.5 μM) of rhodamine B dye was added to one of the streams to distinguish these two streams and thereby to visualize mixing. We here did not demonstrate the mixing of actual analytes or reagents (e.g., deoxyribonucleic acids or other biomolecules). We, however, expect a similar mixing because of strong mixing of background electrolytes, which is demonstrated in the next section.
The first step in experiments was micro/nanochannel cleaning to prevent clogging, especially in nanochannels. We performed the initial flushing step for the just-finished device with 0.1 M sodium hydroxide (NaOH) solution in about 2 min. We took care not to conduct this flushing step too long to protect the oxide layer from NaOH etching. After flushing with NaOH, we flushed the mixer again with deionized (DI) water in about 20 min to remove all the residual NaOH solution. We also performed a cleaning step in the first run of every experiment with a similar procedure, except for a merely 30-second NaOH flushing. After these cleaning procedures, we were ready for production experiments.
We performed mostly combined electroosmotic and pressure-driven flow (EOF + PDF) experiments with only PDF experiments as a control. We first started a pressure-driven flow using syringe pumps. We are able to observe mixing patterns in micro/nanochannel devices using epifluorescent microscopy (Olympus, IX-51) in a dark room condition. The CCD camera (Photometrics, Coolsnap) connected to the microscope took digital images of mixing patterns and exported the visualization data to the computer. We analyzed these collected data using commercial imaging software (Media Cybernetics, Image pro Plus). As discussed below, mixing only with PDF is negligible because the flow rate is too fast compared with spontaneous molecular diffusion. We then turned on electricity to initiate an electroosmotic flow and, more importantly, non-equilibrium electrokinetic phenomena at the micro/nanochannel interfaces. We used a commercial power supply (Itech, IT 6834) to deliver electrical power to the mixing device through platinum wires inserted in the flow streams. The polarity was positive with the same voltage at the two inlets of the device and it is grounded at the outlet.
We characterized our non-equilibrium electrokinetic mixers for different conditions. We varied applied voltages, KCl concentrations, and PDF flow rates using different nanochannel types. Mixing performance was already known to depend on applied voltages and salt concentrations, as reported by Kim et al. (2008). We also found that experimental data with flow rate difference are useful in analyzing how PDF affects mixing capability, especially demonstrating rapid mixing of our device. We also tested four types of nanochannel patterns shown in Fig. 1. These four types are different not only in its unit width but also in their overall width (including spacings) from 130 to 1,900 μm.
3 Results and discussion
3.1 Visualization of vortex generation
Figure 3 shows fluorescent imaging results for (a) a pure pressure-driven flow and (b) a pure electroosmotic flow. Here, we defined zero second (0 s) as the onset time for both types of flows. Figure 3a on the right shows the time-sequence images of a pressure-driven flow. The flow rate was 5 μl/min and the device was the nanochannel type 1 in this case. Each image in the sequence shows two flow streams in two respective microchannel sections. The U-turn of the microchannel is not shown in the figure because it is located below the region shown in the image. The nanochannels are located near the center of each image although they are not clearly visible. These nanochannels connect two microchannel sections. In each image of the sequence, there are a downward stream from the two inlets (left) and a upward stream after making the U turn along the microchannel (right). Only half the stream is bright in downward and upward streams, respectively. Recall that only one inlet reservoir contains rhodamine B while both the inlet reservoirs contain identical KCl solutions. Two clearly distinguished steams indicate that mixing does not occur. The single U turn therefore does not contribute to mixing much from this PDF results.

Fluorescent intensity profiles (left) and fluorescent images (right) for a a pressure-driven flow and b an electroosmotic flow. The coordinate system is defined as the x-axis along the flow and y-axis along the channel width
For a quantitative analysis, we calculated time-averaged fluorescent intensity profiles from all the images. The graph in Fig. 3a on the left shows the variation of the fluorescent intensity I(y) in y direction. We have the coordinate system with x-axis along the flow stream and the y-axis along the channel width. The origin is located where two inlet flow streams merge. We showed the fluorescent intensity profiles at three locations: the uppermost area, the area right ahead of the nanochannel interface, and the area right behind the nanochannel interface. The left half of the curve is close to zero and the right half to one because the right half is the area of a flow stream containing rhodamine B dye. There is a steep slope near the centreline of the channel and it indicates some degree of mixing due to molecular diffusion. If it were a vertical curve with an infinite slope, it would mean perfect non-mixing. The horizontal line would mean perfect mixing. Although three curves with finite slopes indicate some degree of mixing, the mixing performance only with PDF seems very poor.
Figure 3b shows experimental results with a pure electroosmotic flow with an applied voltage of 150 V and 0.5 mM KCl concentration. In Fig. 3b on the right, we observe strong vortex around the nanochannel interface. The time-sequence images show how this vortex structure is changing in time. The images visually indicate mixing behind the nanochannel interface area. We quantified this mixing by calculating the fluorescent intensity [I(y)] profiles, shown in the same figure on the left. The probing locations are similar to the case of the pressure-driven flow. The curves at locations a and b look similar to the corresponding curves in Fig. 3a. This means that there is little mixing ahead of the nanochannel interface for both cases because mixing here should be solely due to slow molecular diffusion. A clear difference from the previous case is found at location c. The curve here is almost horizontal and the fluorescent intensity is nearly uniform. It means that one stream containing a rhodamine B dye and the other stream with no dye are well mixed. It clearly confirmed that strong vortices are generated due to non-equilibrium electrokinetic phenomena at the micro/nanochannel interface.
3.2 Parametric study of mixing performance
We introduce a normalized mixing index I norm obtained from fluorescent intensity data to quantify mixing. We probe a fluorescent intensity I(y) across the width of a microchannel in y direction, as in Fig. 3. From I(y), we calculate a normalized mixing index I norm(x) defined as
Here, I 100% and I 0% indicate a fluorescent intensity for perfect mixing and non-mixing, respectively. The normalized mixing index is a good measure of mixedness at a certain location along the flow stream (x direction). Figure 4 shows the experimental results of pressure-driven and electroosmotic flows each with two different flow rates of 0.1 and 20 μl/min. The device has the nanochannel type 1. We indeed performed two separate experiments with a pure pressure-driven flow and with a combined electroosmotic and pressure-driven flow. We then subtracted the data of PDF from the data of EOF + PDF to extract the data of EOF. The experimental conditions were the applied voltage of 150 V and the KCl concentration of 0.1 mM. The curves for a pure pressure-driven flow show that a mixing index increases slowly downstream. This increase is solely due to molecular diffusion. We can see that mixing is not effective from the small slopes of both curves. The slope for the flow rate of 0.1 μl/min is greater than that for 20 μl/min. With a smaller flow rate, the flow time becomes longer and it results in greater molecular diffusion (and thereby better mixing) at the same location. The data of EOF + PDF and EOF clearly indicate much increased slopes in I norm(x). The curves of EOF + PDF are actually very close to those of EOF for both flow rates. These curves are similar to the curves of PDF right upstream. but they start to deviate from the curves of PDF around x = 190 μm, from where the nanochannels are located. From this point on, the slopes increase very steeply, indicating rapid mixing. The curves of EOF + PDF and EOF asymptote to one around x = 550 μm. It means that mixing is almost completed at this point. Figure 4 clearly shows the effect of non-equilibrium electrokinetic phenomena on mixing performance.

Profiles of normalized mixing index for a pure pressure-driven flow, a pure electroosmotic flow, and a combined pressure-driven and electroosmotic flow
Kim et al. (2008) pointed out two important parameters for mixing performance: an applied voltage and an ion concentration of working fluids. Figure 5a, b shows the performance of our device according to the variations of these parameters. We varied applied voltages between 100 and 230 V in Fig. 5a with the other experimental conditions fixed at 0.1 mM of KCl concentration, 0.1 μl/min of flow rate, and nanochannel type 4. There is a clear trend that mixing completion (the mixing index of unity) occurs further upstream with an increased applied voltage. It means that the mixing length, i.e., the length between I norm = 0 and I norm = 1, becomes shorter with the applied voltage. For a fixed flow rate, which is the case in this experiment, it also means that the mixing time becomes shorter as well. We discuss this mixing time in the next section in more detail. This result of improved mixing performance with a higher applied voltage is consistent with the previous work by Kim et al. (2008). Applying a higher voltage induces stronger non-equilibrium electrokinetic phenomena at the micro/nanochannel interface and thereby improves on mixing performance. Figure 5b shows the results for varying KCl concentrations with the other parameters fixed at 200 V, 0.1 μl/min, and nanochannel type 3. The lower KCl concentration is, the better the mixing performance, which is in good agreement with the work of Kim et al. (2008). The mixing length and mixing time are shortened with a lower concentration. The dependency of mixing upon ion concentration is indebted to the nature of non-equilibrium electrokinetic phenomena: they depend on the nanochannel depth relative to the Debye length, which is a strong function of ion concentration (Kim et al. 2007).

Profiles of normalized mixing index along the fluid flow for variations in a applied voltages, b KCl concentrations, c flow rates, and d nanochannel types
We characterized mixing performance also in terms of flow rates and nanochannel types. Figure 5c shows the mixing index profiles for flow rates between 0.1 and 30 μl/min. The other experimental conditions were 190 V, 0.1 mM KCl, and nanochannel type 4. The profiles show that mixing is poor at a high flow rate. We observed that the vortex structures are stretched towards downstream as the flow rate increases. The mixing region thus becomes wider and the perfect mixing state with I norm = 1 is achieved further downstream. Recall that we observed a similar poor mixing performance at a high flow rate for a pure pressure-driven flow in the previous section. However, mixing performance with an electroosmotic flow is far less sensitive to the flow rate change than with a pure pressure-driven flow. We believe that it is due to the effect of non-equilibrium electrokinetic phenomena. Figure 5d shows the experimental results with four different patterns of nanochannels (see Fig. 1). The overall nanochannel width (including the spacing width) is greater from type 1 to type 4. The other experimental conditions were 190 V, 0.1 mM KCl, and 0.1 μl/min. We initially expected that the type with a wider nanochannel would show a better mixing performance because of a possible larger vortex region The experimental results agree with our initial expectation with the same trend. However, the correlation between the overall width and the mixing performance is not as strong as we initially expected. The size of vortex structures indeed does not change much with the nanochannel overall width.
To further investigate the effects of flow rate, we analysed the experimental data in a different way. We probed the normalized mixing index at a fixed channel center location around x = 600 μm. Figure 6a, b shows the normalized mixing index for varying applied voltages and KCl concentrations, respectively. Both of these figures are consistent with the results in Fig. 5a, b: they confirm the improved mixing performance with a higher voltage and a lower concentration. We also found that the mixing performance is little affected by the flow rate between 0 and 50 μl/min, while we varied the applied voltage from 50 to 250 V and the KCl concentration from 0.1 to 50 mM.

Normalized mixing index at a fixed location for a flow rates and applied voltages under a low applied voltage and b a flow rates and KCl concentrations under a high KCl concentration
In the parametric study of this section, we found that the mixing performance of our device depends strongly on an applied voltage and an ion concentration, whereas it is little affected by a flow rate and a nanochannel width. This insensitiveness on a flow rate could be beneficial for some applications because the device operates effectively with a faster flow condition.
3.3 Characterization of mixing time
Many researchers propose different types of microfluidic mixers these days and their targets are often rapid mixing. The application fields of rapid microfluidic mixers include protein folding, cell activation, drug delivery, nucleic acid sequencing, etc. Researchers try to achieve this goal by varying structures and patterns of channels, different base materials, and diverse active or passive types. We here demonstrate rapid mixing of our device by taking the advantage of non-equilibrium electrokinetic phenomena. We quantified mixing times at different conditions as follows. The mixing time, in principle, should be the time spent between perfect non-mixing (I norm = 0) and perfect mixing (I norm = 1). The experimental data, however, have some errors as well as some fluctuations as in Fig. 5. To take the uncertainty of the data into account, we defined two mixing times, one between I norm = 0.05 and I norm = 0.95 and the other between I norm = 0.10 and I norm = 0.90. We first calculated a mixing length of I norm = [5%, 95%] or [10%, 90%] for all the curves of the last section. We then divided this mixing length by a flow velocity to obtain a mixing time. We can calculate the flow velocity from the pre-set flow rate and the channel cross-sectional area (Depth × Width = 13 μm × 100 μm). In this calculation, we were able to neglect the contribution due to EOF because the estimated EOF flow rate was at least two orders of magnitude lower than the pre-set flow rate. For example, if we use a typical zeta potential of −100 mV, we estimate the EOF flow rate to be 0.015 μL/min at 100 V. This can be compared with the applied flow rate of 30 μl/min fixed in all the experiments.
Figure 7 shows the variation of mixing times in different conditions. The trend of the mixing time profiles is consistent with the mixing index profiles in the previous section: shorter mixing time with an increased applied voltage, a decreased ion concentration, and a wider nanochannel overall width. We achieve the fastest mixing time of about 2 milliseconds (2 ms) with 230 V, 0.1 mM KCl concentration, and nanochannel type 4. This result can be compared with a benchmark case. Mao et al. (2010) demonstrated a chaotic bubble mixer which enables mixing within 20 ms. Ahmed et al. (2009) reported a microfluidic mixer with a single bubble-based acoustic streaming with mixing in 7 ms and Song and Ismagilov (2003) proposed a microfluidic chip with mixing capacity in order of 1 ms using nanoliters of sample. Liau et al. (2005) demonstrated rapid mixing in the order of a few milliseconds with the purpose of mixing crowded biological solutions. All these devices are insensitive to the type of solution to be mixed because they operate based on a physical mechanism. All these devices, however, have rather complex structures often with lengthy channels although this independency is beneficial. They also depend on so-called Peclét number and thereby they depend on flow rate. On the contrary, electrokinetic mixers are simple in design and take a very small space. As a famous previous work, Oddy et al. (2001) showed, there is a micromixer based on electrokinetic instability. They showed that their electrokinetic mixers enabled mixing in as low as 1.5 s using borate and HEPES buffers of 5–250 μS/cm. Their mixer is simple in design like ours albeit with the similar solution-dependency. We believe that our mixer has a potential in applications where solution sensitivity is relatively not important because of the simplicity of design and the relative ease of fabrication. We also think that the relative flow rate-insensitivity of mixer performance could be another benefit of our mixer.

Mixing time against a an applied voltage, b a KCl concentration, and c a nanochannel type
4 Conclusion
We report mixing performance of our non-equilibrium electrokinetic micromixer in this paper. We found that non-equilibrium electrokinetic phenomena clearly contribute to mixing from the experiments with PDF and PDF + EOF. The mixing performance depends on several parameters: applied voltage, ion concentration, flow rate, and nanochannel width. We quantified this performance using a mixing index. The mixing index profiles show that more efficient micromixing is achieved with increased applied voltage, decreased ion concentration, and wider nanochannel overall width. Mixing performance depends strongly on the change in the applied voltage and the ion concentration, whereas it is rather insensitive to the change in the other two parameters. We observed the same trend in mixing time results. The best performance of the device was 2 millisecond-mixing with the highest voltage and lowest ion concentration. We believe that the non-equilibrium electrokinetic mixer has several advantages as an efficient mixing device in some applications. Those include simple design, small size, relatively easy fabrication, and DC voltage, which is readily available in most cases. Although the mixing performance is affected by the solution concentration, its relative insensitivity to flow rate could be another benefit.
References
Ahmed D, Mao X, Shi J, Juluri BK, Huang TJ (2009) A millisecond micromixer via single-bubble-based acoustic streaming. Lab Chip 9:2738–2741
Chen C, Cho C (2008) A combined active/passive scheme for enhancing the mixing efficiency of microfluidic devices. Chem Eng Sci 63:3081–3087
Chen J, Yang R (2007) Electroosmotic flow mixing in zigzag microchannels. Electrophoresis 28:975–983
Choi YS, Kim SJ (2009) Electrokinetic flow-induced currents in silica nanofluidic channels. J Colloid Interface Sci 333:672–678
Di Carlo D (2009) Inertial microfluidics. Lab Chip 9:3038–3046
Dukhin SS (1993) Nonequilibrium electric surface phenomena. Adv Colloid Interface Sci 44:1–134
Egawa T, Durand JL, Hayden EY, Rousseau DL, Yeh S (2009) Design and evaluation of a passive alcove-based microfluidic mixer. Anal Chem 81:1622–1627
El Moctar AO, Aubry N, Batton J (2003) Electro-hydrodynamic micro-fluidic mixer. Lab Chip 3:273–280
Erbacher C, Bessoth FG, Busch M, Verpoorte E, Manz A (1999) Towards integrated continuous-flow chemical reactors. Mikrochim Acta 131:19–24
Frommelt T, Kostur M, Wenzel-Schaefer M, Talkner P, Haenggi P, Wixforth A (2008) Microfluidic mixing via acoustically driven chaotic advection. Phys Rev Lett 100:034502
Gadish N, Voldman J (2006) High-throughput positive-dielectrophoretic bioparticle microconcentrator. Anal Chem 78:7870–7876
Gambin Y, Simonnet C, VanDelinder V, Deniz A, Groisman A (2010) Ultrafast microfluidic mixer with three-dimensional flow focusing for studies of biochemical kinetics. Lab Chip 10:598–609
Goet G, Baier T, Hardt S (2009) Micro contactor based on isotachophoretic sample transport. Lab Chip 9:3586–3593
Haeberle S, Zengerle R (2007) Microfluidic platforms for lab-on-a-chip applications. Lab Chip 7:1094–1110
Hardt S, Drese KS, Hessel V, Schonfeld F (2005) Passive micromixers for applications in the microreactor and mu TAS fields. Microfluid Nanofluid 1:108–118
Hardt S, Drese KS, Hessel V, Schonfeld F (2006) Theoretical and experimental characterization of a low-Reynolds number split-and-recombine mixer. Microfluid Nanofluid 2:237–248
Hessel V, Lowe H, Schonfeld F (2005) Micromixers—a review on passive and active mixing principles. Chem Eng Sci 60:2479–2501
Hirschberg S, Koubek R, Moser F, Schoeck J (2009) An improvement of the Sulzer SMX (TM) static mixer significantly reducing the pressure drop. Chem Eng Res Des 87:524–532
Hsiung S, Lee C, Lin J, Lee G (2007) Active micro-mixers utilizing moving wall structures activated pneumatically by buried side chambers. J Micromech Microeng 17:129–138
Kim SJ, Wang Y, Lee JH, Jang H, Han J (2007) Concentration polarization and nonlinear electrokinetic flow near a nanofluidic channel. Phys Rev Lett 99:044501
Kim D, Raj A, Zhu L, Masel RI, Shannon MA (2008) Non-equilibrium electrokinetic micro/nanofluidic mixer. Lab Chip 8:625–628
Kim SJ, Li LD, Han J (2009) Amplified electrokinetic response by concentration polarization near nanofluidic channel. Langmuir 25:7759–7765
Kim SJ, Song YA, Han J (2010) Nanofluidic concentration devices for biomolecules utilizing ion concentration polarization: theory, fabrication, and applications. Chem Soc Rev 39:912–922
Lee H, Voldman J (2007) Optimizing micromixer design for enhancing dielectrophoretic microconcentrator performance. Anal Chem 79:1833–1839
Lee CY, Lee GB, Fu LM, Lee KH, Yang RJ (2004) Electrokinetically driven active micro-mixers utilizing zeta potential variation induced by field effect. J Micromech Microeng 14:1390–1398
Liau A, Karnik R, Majumdar A, Cate JHD (2005) Mixing crowded biological solutions in milliseconds. Anal Chem 77:7618–7625
Lund-Olesen T, Buus BB, Howalt JG, Hansen MF (2008) Magnetic bead micromixer: influence of magnetic element geometry and field amplitude. J Appl Phys 103:07E902
Mani A, Zangle TA, Santiago JG (2009) On the propagation of concentration polarization from microchannel-nanochannel interfaces. Part I: Analytical model and characteristic analysis. Langmuir 25:3898–3908
Mansur EA, Mingxing Y, Yundong W, Youyuan D (2008) A state-of-the-art review of mixing in microfluidic mixers. Chin J Chem Eng 16:503–516
Mao P, Han JY (2005) Fabrication and characterization of 20 nm planar nanofluidic channels by glass–glass and glass–silicon bonding. Lab Chip 5:837–844
Mao X, Juluri BK, Lapsley MI, Stratton ZS, Huang TJ (2010) Milliseconds microfluidic chaotic bubble mixer. Microfluid Nanofluid 8:139–144
Miller EM, Wheeler AR (2008) A digital microfluidic approach to homogenenous enzyme assays. Anal Chem 80:1614–1619
Min J, Kim JH, Kim S (2004) Microfluidic device for bio analytical systems. Biotech Bioprocess Eng 9:100–106
Neils C, Tyree Z, Finlayson B, Folch A (2004) Combinatorial mixing of microfluidic streams. Lab Chip 4:342–350
Nguyen NT, Wu ZG (2005) Micromixers—a review. J Micromech Microeng 15:R1–R16
Niu XZ, Liu LY, Wen WJ, Sheng P (2006) Active microfluidic mixer chip. Appl Phys Lett 88:153508
Oddy MH, Santiago JG, Mikkelsen JC (2001) Electrokinetic instability micromixing. Anal Chem 73:5822–5832
Park SY, Russo CJ, Branton D, Stone HA (2006) Electrokinetic instability micromixing. J Colloid Interface Sci 297:832–839
Park S, Lee S, Moon JH, Yang S (2009) Holographic fabrication of three-dimensional nanostructures for microfluidic passive mixing. Lab Chip 9:3144–3150
Plecis A, Nanteuil C, Haghiri-Gosnet A, Chen Y (2008) Electropreconcentration with charge-selective nanochannels. Anal Chem 80:9542–9550
Pu QS, Yun JS, Temkin H, Liu SR (2004) Ion-enrichment and ion-depletion effect of nanochannel structures. Nano Lett 4:1099–1103
Puleo CM, Wang T (2009) Microfluidic means of achieving attomolar detection limits with molecular beacon probes. Lab Chip 9:1065–1072
Ridgeway WK, Seitaridou E, Phillips R, Williamson JR (2009) RNA-protein binding kinetics in an automated microfluidic reactor. Nucleic Acids Res 37:e142
Roder H, Maki K, Cheng H, Shastry MCR (2004) Rapid mixing methods for exploring the kinetics of protein folding. Methods 34:15–27
Rubinshtein I, Zaltzman B, Pretz J, Linder C (2002) Experimental verification of the electroosmotic mechanism of overlimiting conductance through a cation exchange electrodialysis membrane. Russ J Electrochem 38:853–863
Rubinstein SM, Manukyan G, Staicu A, Rubinstein I, Zaltzman B, Lammertink RGH, Mugele F, Wessling M (2008) Direct observation of a nonequilibrium electro-osmotic instability. Phys Rev Lett 101:236101
Sasaki N, Kitamori T, Kim HB (2006) AC electroosmotic micromixer for chemical processing in a microchannel. Lab Chip 6:550–554
Sasaki N, Kitamori T, Kim H (2010) Experimental and theoretical characterization of an AC electroosmotic micromixer. Anal Sci 26:815–819
Schulte TH, Bardell RL, Weigl BH (2002) Microfluidic technologies in clinical diagnostics. Clinica Chimica Act 321:1–10
Shcherbakova I, Brenowitz M (2008) Monitoring structural changes in nucleic acids with single residue spatial and millisecond time resolution by quantitative hydroxyl radical footprinting. Nat Protoc 3:288–302
Song H, Ismagilov RF (2003) Millisecond kinetics on a microfluidic chip using nanoliters of reagents. J Am Chem Soc 125:14613–14619
Stroock AD, Dertinger SKW, Ajdari A, Mezic I, Stone HA, Whitesides GM (2002) Chaotic mixer for microchannels. Science 295:647–651
Tofteberg T, Skolimowski M, Andreassen E, Geschke O (2010) A novel passive micromixer: lamination in a planar channel system. Microfluid Nanofluid 8:209–215
Villermaux E, Stroock AD, Stone HA (2008) Bridging kinematics and concentration content in a chaotic micromixer. Physical Review E 77:015301
Wang Y, Zhe J, Dutta P, Chung BT (2007) A microfluidic mixer utilizing electrokinetic relay switching and asymmetric flow geometries. J Fluids Eng Trans ASME 129:395–403
Wu Z, Li D (2008) Mixing and flow regulating by induced-charge electrokinetic flow in a microchannel with a pair of conducting triangle hurdles. Microfluid Nanofluid 5:65–76
Yan D, Yang C, Miao J, Lam Y, Huang X (2009) Enhancement of electrokinetically driven microfluidic T-mixer using frequency modulated electric field and channel geometry effects. Electrophoresis 30:3144–3152
Yaralioglu GG, Wygant IO, Marentis TC, Khuri-Yakub BT (2004) Ultrasonic mixing in microfluidic channels using integrated transducers. Anal Chem 76:3694–3698
Yi CQ, Li CW, Ji SL, Yang MS (2006) Microfluidics technology for manipulation and analysis of biological cells. Anal Chim Acta 560:1–23
Yossifon G, Chang H (2008) Selection of nonequilibrium overlimiting currents: universal depletion layer formation dynamics and vortex instability. Phys Rev Lett 101:254501
Zaltzman B, Rubinstein I (2007) Electro-osmotic slip and electroconvective instability. J Fluid Mech 579:173–226
Acknowledgement
This work was supported by the Basic Science Research Program (Grant No. 2011-0009993) through the National Research Foundation of Korea (NRF) funded by the Ministry of Education, Science and Technology.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Lee, S.J., Kim, D. Millisecond-order rapid micromixing with non-equilibrium electrokinetic phenomena. Microfluid Nanofluid 12, 897–906 (2012). https://doi.org/10.1007/s10404-011-0918-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10404-011-0918-y