
Abstract
A novel method for fabricating nano- or submicro-fluidic PMMA chips using photoresist-free UV-lithography and UV-assisted low-temperature bonding was developed. The nano- or submicro-channels were fabricated by exposing the PMMA substrate to the UV-light through a mask for a certain time. The PMMA substrate with channels and another flat PMMA cover sheet were pretreated with the UV-light for 2 h before they were brought together in running water. The bonding was carried out under a pressure of (1.19 ± 0.12) × 105 Pa and at a temperature of 45°C for 35 min. The chips bonded in this way could bear a tensile of 6.71 ± 2.50 MPa, and the deformation of the bonded channel was about 13%. A hybrid micro- and nano-fluidic PMMA chip fabricated with the developed method was demonstrated for the test of the electrokinetically driven ion enrichment and ion depletion.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
Nanofluidic chips have become expanding attractive for the applications in the chemical and biochemical analysis due to the unique properties of the nanochannels. The special character of nanofluidics arises from the fact that the channel dimension is comparable to the thickness of electric double layer (EDL) and the size of biomolecules such as DNAs and proteins. Consequently, any changes in the channel dimension or the thickness of EDL can significantly affect mass transport of charged species inside the nanochannels. Thus, various nanofluidic devices have been developed for fluid manipulation, analyte concentration, and separation (Abgrall and Nguyen 2008; Kovarik and Jacobson 2009; Gardeniers 2009; Kim et al. 2010; Napoli et al. 2010). For example, proteins can be enriched up to million folds via the electrokinetic concentration polarization occurred on nanofluidic filters (Wang et al. 2005; Wu and Steckl 2009; Kim et al. 2006). DNAs can be stretched when they are transported through nano-scale conduits (Yang et al. 2007; Krishnan et al. 2007). Furthermore, without using a gel matrix, DNAs can be separated with a nanofluidic device containing entropic traps (Han and Craighead 2000; Cabodi et al. 2002) or with nanocapillaries (Wang et al. 2008).
One critical issue in nanofluidics is the fabrication of nanofluidic devices with simple, reliable, and cost-effective strategies. Usually, fabrication of nanofluidic devices involves two key steps, preparation of nanostructures on a substrate and sealing of the nanostructure on the substrate with a cover sheet. As to the fabrication of nanochannels, many dry etching techniques, such as reactive ion etching (Mao and Han 2005), electrobeam lithography (Yasin et al. 2001), focused-ion beam lithography (Cannon et al. 2004), proton beam writing (Mahabadi et al. 2006), interferometric lithography (O’Brien et al. 2003), and scanning probe lithography (Ginger et al. 2004), have been reported to produce the nanostructures on various substrates. These techniques enable precise fabrication of 1-D, 2-D, and even 3-D nanostructures. However, they are cost-expensive and difficult to scale-up for mass production. In many cases, fluidic chips with 1-D nanofluidic channels, which keep channel depth in nanometer range (1–100 nm) while have the width in the micrometer scale and the length at a millimeter or centimeter level, are needed. Under these circumstances, simple alternatives for such nanolithography-based techniques would be practical to fabricate “shallow” rather than “narrow” channels. One of such alternatives is the UV-lithography and wet etching technique for fabrication of nanostructures on glass substrate (Mao and Han 2005; He et al. 2008). Several reviews have been recently published on the fabrication of nanofluidic devices (Mijatovic et al. 2005; Perry and Kandlikar 2006; He and Chen 2008; Gates et al. 2005).
Compared with silicon, quartz, and glass, polymeric materials offer substantially lower material costs and chip-manufacturing costs as well. Thus, polymer substrates represent an attractive alternative for an eventual marketing of micro- and nano-fluidic devices. Apart from the aforementioned dry etching techniques that can be used to directly prepare nanostructure on thermoplastic polymer sheets, molding and embossing techniques are the mostly widely adopted methods to prepare nanostructures on polymer substrates. With these techniques, molds with nanometer relief are inevitably prepared with complicated technology. Recently, Chantiwas et al. (2010) developed a simple method to prepare nanofluidic structure on polymethyl methacrylate (PMMA) sheets via thermo-embossing the substrates with an optical mask serving as a mold. The optical mask consisted of a sodalime glass plate with a thin Cr layer (~100 nm, defined the nanoslit depth) that was patterned using standard UV-lithography and wet etching. Mechanical stretching (Sivanesan et al. 2005) and electrical breakdown (Lee et al. 2007; Yu et al. 2008) have also been reported to prepare nanochannels or nanogaps on polymer substrates. Using these two techniques, however, the inner diameter of the nanochannels can hardly be accurately controlled.
Bonding of the nanochannels is also challenging due to the problem of nanochannel collapse. For polymer microfluidic devices, thermo-bonding is most widely used to bond the substrate with channel structures and a flat cover sheet. It is based on the fusion of the intimately contacted polymer surfaces under the conditions of a high pressure and at a temperature higher than or close to the polymer’s glass transition T g (Tsao and DeVoe 2009). When this technique is applied to seal nanochannels, it will lead to nanostructure collapse. Thus, novel strategies of bonding polymeric nanochannels at low temperatures, such as oxygen-plasma-assisted low-temperature bonding (Chantiwas et al. 2010; Abgrall et al. 2007), microwave-assisted bonding (Lei et al. 2004), and solvent-assisted sealing (Cho et al. 2010), have been reported.
In this article, we present a novel and low-cost method for fabricating PMMA fluidic chips with 1-D nanochannels. The nanostructures were photoresist-free UV-lithographied onto PMMA substrates with the UV-light emitted from a low-pressure mercury lamp. UV pre-treatment of both the substrate containing channel network and the flat cover sheet generated on the PMMA surfaces hydrophilic moieties that helped the bonding of the PMMA substrate and the cover sheet at a low temperature and under a low pressure. With the developed method, fabrication of the PMMA fluidic chips containing 1-D nanochannels can be conducted in common wet chemistry labs without access to a clean room and expensive facilities. A hybrid micro- and nano-fluidic PMMA chip fabricated with the present method was demonstrated for the electrokinetically driven ion enrichment–depletion.
2 Experimental
2.1 Chemicals and apparatus
PMMA sheets of 3.0 mm thickness were purchased from Jinxi Research Institute of Chemical Industry (Huludao, Liaoning, China). Fluorescein isothiocyanate isomer (FITC) was from sigma (St. Louis, MO, USA).
A low-pressure mercury lamp originally used as the source of ultraviolet analyzer was employed for the UV-lithography and UV-pretreatment of the PMMA sheets. A model GY-6 high-voltage supply (6 kV, North-eastern University, Shenyang, China) under the control of a computer was used for the test of the electrokinetic ion enrichment–depletion. A model Eclipse TE2000-S fluorescent microscopy (Nikon, Japan) equipped with a CCD camera was adopted to collect the fluorescent images.
2.2 Fabrication of nanochannels on PMMA via photoresist-free UV-lithography
A PMMA substrate of 30 × 50 × 3 mm was cleaned with ethanol and water, and then dried in a nitrogen stream. The cleaned PMMA was exposed, in the air, to the UV-light emitted from a low-pressure mercury lamp (the radiation power at 254 nm was measured 2.07 mW cm−1, and ozone generation was accompanied) through a quartz glass photomask (Kong et al. 2007) with a designed pattern for a specified time (depending on the required depth of the nanochannels). The pattern of nanochannels was lithographied onto the PMMA substrate due to the ozone-assisted UV ablation (Fig. 1a).

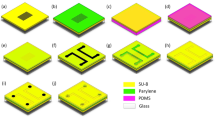
Schematic of the fabrication procedure for fluidic PMMA chips with nano- or submicro-channels. a the PMMA substrate was exposed to the UV-light for a certain time through a photomask to etch the nanochannels; b the PMMA substrate with the nanochannels and the flat PMMA cover sheet were irradiated with the UV-light for 2 h for surface modification; c the UV-pretreated PMMA sheets were brought into contact in running water; d the two PMMA sheets were sandwiched with two pieces of PDMS and two pieces of glass plates, after which the assembly was clamped with four clamps and put into an oven for bonding
2.3 Bonding of the nanofluidic PMMA chip
The PMMA substrate with the channel network and a flat PMMA cover sheet were exposed, in the air, to the UV-light for 2 h (Fig. 1b). Then, the two UV-treated PMMA sheets were brought into conformable contact in running deionized water (Fig. 1c). The contacted PMMA sheets were then sandwiched by two pieces of PDMS and two pieces of glass (Fig. 1d). The assembly was clamped with four clamps that produced a pressure of (1.19 ± 0.12) × 105 Pa, and the assembly was incubated in an oven set at 45°C for 35 min.
2.4 Fabrication of the hybrid micro- and nano-fluidic PMMA chip
The hybrid micro- and nano-fluidic PMMA chip used for the test of the electrokinetic ion enrichment–depletion consisted of an upper PMMA substrate containing two U-shaped microchannels connected by a nanogap and a lower flat PMMA cover sheet. The U-shaped microchannels were first thermo-embossed (at 108°C and under 1.33 MPa) onto the PMMA substrate with a silicon mold. Afterward, the nanogap connecting the U-shaped microchannels was UV-lithographied onto the substrate as described in Sect. 2.2. The bonding process was the same as that described in Sect. 2.3.
2.5 Characterization
2.5.1 Water contact angle measurement
A model Powereach JC2000C3 contact angle goniometer (Zhongchen Co, Shanghai, China) was used to measure the contact angle of deionized water on the PMMA surface. A sample PMMA sheet was placed on the measuring platform of the contact angle goniometer, which was set at 25°C. Approximately, 10 μl of deionized water was dropped onto the surface of the PMMA with a hand-driven peristaltic pump. After 20 s, the image of the water droplet on the PMMA surface was taken with the goniometer-equipped camera. The contact angle was then measured using the software provided by the manufacturer.
2.5.2 ATR-FT-IR spectra
A model Nexus-670 attenuated total reflection Fourier transform infrared spectrometer (Nicolet, USA) was used to characterize the surface chemistry of PMMA sheets before and after UV-treatment. The ATR-FT-IR spectra for native and UV-treated PMMA sheets were recorded from 400 to 4000 cm−1 at a 4 cm−1 resolution under a dry condition. All PMMA samples were heated in an oven to remove adsorbed water moisture immediately before they were subjected to ATR-FT-IR analysis.
2.5.3 Electroosmotic flow
Electroosmotic flows (EOFs) supported by the native and UV-treated PMMA micro channels were measured with the electric current method (Huang et al. 1988) with phosphate buffers (pH 7.0, 10 mM for the concentrate buffer and 9 mM for the dilute one) under the electric filed of 110 V cm−1.
2.5.4 Tensile measurement
A model of WDW-100 (Jinan Shijin Croup Corp, Jinan, China) electronic universal testing machine was used to evaluate the bonding strength of the PMMA chips. Two 2 cm × 2 cm × 3 mm PMMA sheets were bonded as described in Sect. 2.3. The bonded PMMA sample was carefully glued to two 2 cm × 2 cm × 5 mm steel bolts with Epoxy. After curing, the entire set-up was loaded into the self-tightening grippers of the electronic universal testing machine to conduct the tensile measurement. During the experiments, the grippers were separated at a speed of 1 mm min−1.
2.5.5 Surface metrology
A stylus profilometer (Dektak 150, Veeco Inst. Inc, USA) was employed to measure the profile of the nanochannels. A sample PMMA sheet was placed on the platform of the profilometer, and then the stylus was down to the surface of the PMMA. Afterward, the position of the PMMA was adjusted to ensure the nanochannel being perpendicular to the scanning trace of the stylus before the stylus began to scan. Scanning rate was set at 2.5 μm s−1. The data were processed with the software provided by the manufacturer.
Atomic force microscope (AFM) images of the UV-lithographied channels were collected in tapping mode using a Veeco Multiple SPM with Nanoscope IVa controller (Veeco co, USA). Surface roughness expressed in root mean square (RMS) was calculated by the software provided by the manufacturer.
2.6 Procedure of the electrokinetic ion enrichment–depletion
The channel network was sequentially flushed with deionized water and the running buffer (50 μM borate buffer), and finally filled with a solution of 20 μM FITC in 50 μM borate buffer. A voltage of 1.0 kV was applied to the right reservoirs while the left ones grounded. The enrichment–depletion process was observed with a florescence microscope and recorded with the microscope-equipped CCD camera.
3 Results and discussion
3.1 Preparation of nano- or submicro-channels on PMMA sheets
McCarley et al. (2005) reported that PMMA and polycarbonate (PC) surface underwent photolysis when exposed to the UV-light emitted from the low-pressure mercury lamp, resulting in the formation of some hydrophilic moieties on the UV-exposed region. Our previous study (Kong et al. 2006) reported that PC sheets could be UV-lithographied to a depth of 130 nm using the low-pressure mercury lamp as the source. Haiducu et al. (2008) also reported that exposure of PMMA sheets to UV-light from a low-pressure mercury lamp resulted in an etched pattern, the depth of which could be developed in isopropanol–water developing solution to decades of micrometers. They (Johnstone et al. 2008) ascribed the etched depth to the physical shrinkage of the polymer due to the increase in the free volume of the PMMA chains after UV-exposure. In the present study, we observed that after PMMA sheets were exposed to the UV-light through a photomask, not only the hydrophilic moieties were formed on the UV-exposed region (see the discussion in Sect. 3.2.1) but also recessions with the depth of nanometer to sub-micrometer levels were produced on the UV-exposed region, and the depth of the recessions increased with the exposed dose of the UV-light. In other words, 1-D nano patterns could be dryly etched onto PMMA sheets with the photoresist-free UV-lithography. Figure 2 showed the influence of UV-exposed time on the etched depth of 1-D straight nanochannels prepared with such a UV-lithography. As can be seen in the figure, the channel depth increased with the UV-exposed time, with the depth-time profile fitting into a quadric polynomial curve. Obviously, the recessions or nanochannels were formed due to the photo-ablation effect of the UV-light, which have been previously reported by others to machine polymer surfaces such as PMMA, PC, etc. (Srinivasan and Lazare 1985; Zhang et al. 1994; Lippert and Dickinson 2003). It should be pointed out that the low-pressure mercury lamp that emitted the UV-light in company with the generation of ozone (O3) was much more effective for the UV-lithography than the low-pressure mercury lamp that did not generate ozone. This might be that the former emitted not only the UV-light of 254 nm but also the UVD-lights of 185 nm that could be absorbed by oxygen in the air, resulting in the generation of ozone and atomic oxygen that acted as a strong oxidizing agent (Tsao et al. 2007). The strong oxidizing reagent may assist to dryly etch the polymer surfaces. In the following study, the low-pressure mercury lamp that can generate ozone was used.

The effect of the UV-exposed time on the depth of UV-lithographied channels. The black squares with error bars represent the measured data, while the thin line is a quadric polynomial fitting curve. The power of the UV-light was measured to be 2.07 mW cm−1 at 254 nm
It was also noticed that the depth of UV-lithographied nanochannels increased by around 20% after they were vigorously flushed with water. This would most possibly be due to that the non-volatile molecular fragments of the UV-decomposed polymer chains deposited on bottom of the nanochannels were flushed away from the nanochannels. Figure 3 shows the AFM images for a section of the nanochannel prepared with the UV-lithography. As shown in Fig. 3, the roughness of the masked PMMA surface was rather smooth, RMS measured to be about 4 nm. At the nanochannel bottom (UV-exposed region), the surface roughness was almost the same (4 nm) as the UV-non-exposed region if the UV-lithography was conducted for 1 h (Fig. 3a), and the roughness remained basically unchanged after the etched channel was water flushed (Fig. 3c) indicating that the smooth PMMA surface was not significantly damaged for a low dose exposure (about 7.5 J cm−2) UV-lithography. Nevertheless, if the UV-lithography time increased to 3 h, equivalent to UV dose of 22.4 J cm−2, the surface roughness at the channel bottom increased to a RMS of 9 nm (Fig. 3b), and which further increased to about 20 nm after water flushing (Fig. 3d), meaning that measurable surface damage occurred due to the high dose UV-exposure.

AFM images of partial nanochannels prepared by the UV-lithography. UV-lithography time of 1 h for (a) and (c), and 3 h for (b) and (d). Images of (a) and (b) were taken before water flushing, and those of (c) and (d) were taken after water flushing. The scan ranges were 20 × 20 μm. The Z-range was 600 nm for (a) and (c), and 1400 nm for (b) and (d). Image data: Height
For the fabrication of hybrid micro and nano channels on a PMMA substrate, two steps were required as described in Sect. 2.4. In the first step, micro channels were thermo-embossed onto the substrate with an appropriate mold. In the second step, the nanochannel was UV-lithographied onto the substrate through a photomask that had been aligned to the substrate containing the embossed microchannels.
3.2 UV-assisted bonding
Recently, Tsao et al. (2007) developed an UV/O3-assisted low-temperature bonding method for bonding PMMA and cyclic olefin copolymer (COC) microfluidic chips. In their method, bonding was conducted for 10 min at the temperature of 60°C and under the pressure of 4.8 MPa. The authors claimed that the developed method caused no measureable channel deformation for a PMMA microchannel of 500 μm in width and 180 μm in depth. When this method was directly adopted to bond the PMMA chips with nanochannels in our preliminary tests; however, the nanochannels were completely collapsed. Thus, in this study, UV-assisted low-temperature bonding was further investigated to satisfy the needs of bonding PMMA chips with nanochannels.
3.2.1 Characterization of UV-treated PMMA surface
The surface chemistry of the UV-treated PMMA surfaces was characterized with water contact angle, EOF, and ATR-FT-IR spectra. Figure 4 showed the water contact angles on the surface of the PMMA sheets with varied UV-exposure time. The water contact angles decreased rapidly from 71 ± 2° to 23 ± 2° within 1.5 h, and then leveled off. In addition, EOF mobility supported by the microchannel made of native PMMA was (2.95 ± 0.30) × 10−4 cm2 V−1 s−1, while that supported by the UV-treated microchannel was (4.85 ± 0.21) × 10−4 cm2 V−1 s−1. Both the contact angle and EOF data were quite similar to what the previous studies reported (Wei et al. 2005). These observations indicated that UV-irradiation turned the PMMA surface from hydrophobic to hydrophilic due to the photo-generation of hydrophilic functionalities on the UV-exposed surface of PMMA.

The effect of the UV-exposed time on the water contact angles on the PMMA surface
ATR-FT-IR spectra confirmed the changes in surface chemistry after the PMMA sheets were subjected to the UV-irradiation. As shown in Fig. 5, one major difference between the ATR-FT-IR spectrum of the native PMMA and that of the UV-treated PMMA lies in the changes of the absorption peaks of the carbonyl groups. The absorption peak at 1730 cm−1, which can be assigned to the carbonyl stretching vibration of the side ester group, decreased after UV-irradiation. Simultaneously, the whole carbonyl band was broadened, indicating that new oxidized groups were formed due to the photochemical reaction. The broadened shoulder band appearing near 1720–1670 and 1800–1750 cm−1 (pointed by arrows in Fig. 5) can be respectively attributed to aliphatic ketones, and to peracid and perester groups (Kaczmarek et al. 2000). In company of the decrease in the peak height of 1730 cm−1 for the UV-treated PMMA, a broaden absorption band appeared at 3650–3150 cm−1, which could be assigned to the O–H stretching vibration of the hydroxyl group or peroxide group. Based on these significant changes, we presumed that hydrophilic groups such as hydroxyl and carboxylic acid moieties were formed on UV-exposed PMMA surfaces.

ATR-FT-IR spectra of the native PMMA (the solid line) and the UV-treated PMMA (the dotted line)
Since the UV-treatment of the PMMA surfaces was conducted after the nanochannels had been UV-lithographied on the PMMA substrates, the effect of the secondary UV-irradiation on the depth of the nanochannels was examined. It was found that the nanochannels were deepened by 40–70% after the bulk surface was secondarily irradiated by the UV-light for 2 h, depending on the original depth of the UV-lithographied nanochannels. It can be understood if one takes it in mind that the depth of the nanochannels was increased with the UV-exposing time in the way of quadratic polynomial. Thus, the UV-dose of secondary UV-treatment would produce deeper etching of the polymer in the region of UV-lithographied nanochannels than in the other region that had never been exposed to the UV-light; consequently, the depth of the nanochannels was deepened during the secondary UV-treatment.
3.2.2 Bonding procedures
Initially, immediately after the PMMA substrate with the nanochannel network and the flat PMMA cover sheet were UV-treated, the two sheets were brought into contact in air, sandwiched with two PDMS slabs and two glass plates, clamped with four clamps that produced a pressure of (1.54 ± 0.14) × 105 Pa, and left at room temperature for 5 min. With this dry-bonding procedure (termed as dry bonding due to no water was involved), the two PMMA sheets could be bonded with quite good strength. Pull-test measurements showed that a tensile of 2.22 ± 0.29 MPa could be tolerated for the dry-bonded sheets. In contrast, if the two PMMA sheets without UV-treatment were bonded at the same conditions [room temperature, (1.54 ± 0.14) × 105 Pa, 5 min]; the stuck sheets were delaminated within several minutes after the pressure was released. One likely reason for these observations is that the improved wettability of the UV-treated PMMA sheets enables more intimate contact between the surfaces, which facilitates the inter diffusion of short polymer chains generated via photochemical chain scission, consequently improves the bonding strength. In addition, the formation of hydrogen bonds between the intimately contacted PMMA surfaces via the hydroxyl and carboxyl groups might also be helpful to strengthen the bonding of the UV-treated PMMA sheets. With this procedure, unfortunately, more or less air bubbles would remain in-between the two layers of the PMMA sheets, because a few air dusts left on the surfaces of the sheets prevented them from forming intimate contact of the whole surfaces.
To solve the problem, an improved wet bonding procedure was developed. With this method, two UV-treated PMMA sheets were brought into conformable contact under the running water (see Fig. 1c) as Jia et al. (2004) did for bonding glass microfluidic chip at room temperature. Contact of the two sheets under the running water could keep the air and dust away from the surfaces of the PMMA on one hand. On the other hand, leaving a thin water film between the PMMA sheets. This water film might be beneficial to the hydrogen bond formation between the two PMMA sheets. After clamped with four clamps that generated a pressure of (1.19 ± 0.12) × 105 Pa, the assembly was incubated in an oven. The incubation temperature and time were optimized within the ranges of 45–75°C and 20 min to 3 h. Incubated at 45°C for 35 min, the water film between the two sheets was dried, and the sheets were perfectly bonded, no air bubbles remaining in-between the two sheets. Under these conditions, moreover, the bonded nanochannels suffered minimum risk of being partially collapsed. Such bonded chips can bear a tensile of 6.71 ± 2.50 MPa, significantly higher than that (2.22 ± 0.29 MPa) obtained with the above mentioned UV-assisted dry-bonding procedure. The bonding strength was also twice-fold higher than that (2.67 ± 0.54 MPa) offered by the conventional thermal bonding conducted at 90°C and 2.9 MPa, which made the nanochannels completely collapsed.
3.3 Channel deformation
It has been reported that the smaller the aspect (depth-to-width) ratio of nanochannels is, the more susceptible the channels are to be deformed or collapsed during bonding (He et al. 2008). For the bonding of nanofluidic PMMA chips with the present wet bonding procedure (see Sect. 2.3), the nanochannels suffered the risk of being partially collapsed if the aspect ratio of the nanochannels was less than 1 × 10−3. Thus, for a 1-D nanochannel with the expected depth of about 100 nm, the width of the channel should not exceed to 100 μm. Taking the nanochannels of 100 nm in depth and 90 μm in top width (aspect ratio of 1.1 × 10−3) as example, the extent of the channel deformation was evaluated via measuring the profiles of the channels before and after being subjected to an analogical wet bonding procedure. The term of analogical wet bonding means that the PMMA substrate with the nanochannels and the flat PMMA cover sheet were bonded under the same condition as the wet bonding procedure described in the above section except that both the substrate and the cover sheet were not subjected to UV-treatment before bonding. Without the UV-treatment, the bonded chip could be easily delaminated and the re-opened substrate and cover sheet had a smooth surface. Otherwise, the re-opened surface would be very rough so that the profile of nanochannels could not be properly measured with a stylus profilometer. It was observed that after the analogical wet bonding the channel depth decreased by (13 ± 9)% (mean ± SD, n = 3) in comparison to the water-flushed channels. In addition, no relief of the channel pattern was observed by scanning the re-opened cover sheet. Figure 6 shows the typical profiles of a nanochannel measured before and after the analogical wet bonding. Figure 7 is a fluorescence image of an array of nanochannles of 100 nm in lithographied depth, which had been bonded with the wet bonding procedure as described in Sect. 2.3.

Typical channel profiles measured with an α-profilemeter after the nanochannel was UV-lithographied (a), and the lithographied channel was flushed with water (b), and the water-flushed channel was subjected to analogical bonding process and re-opened again (c). Conditions for the analogical wet bonding: both the channel-structured PMMA substrate and a flat PMMA sheet were brought into contact under running water, and the contacted sheets were bonded at the pressure of (1.19 ± 0.12) × 105 Pa and the temperature of 45°C for 35 min

A fluorescence image of the PMMA nanochannels bonded at the pressure of (1.19 ± 0.12) × 105 Pa and the temperature of 45°C for 35 min after the PMMA sheets were UV-treated for 2 h. A solution of 1 mM FITC in 10 mM borate buffer (pH 9.2) was filled in the nanochannels
3.4 Application
The hybrid micro- and nano-fluidic PMMA chip schematically shown in Fig. 8a was applied for the electrokinetic enrichment and depletion (Pu et al. 2004) of FITC dye anions. As shown in Fig. 8b, dim but uniform fluorescence was observed in both of the microchannels (no fluorescence was observed in the nanogap because the depth of the gap was too shallow). Once a voltage of 1.0 kV was applied to the reservoirs of the right-handed micro channel while the reservoirs of the left-handed grounded, the fluorescent intensity near the cathode end of the nanogap quickly increased meanwhile that near the anode end decreased (Fig. 8c and d), the typical concentration polarization phenomenon caused by the ion-selective permeation through a nanochannel. (note, the surface of the PMMA was negatively charged at pH 8.2 as the EOF was directed to cathode). These experimental results verified that the nanogap did possess one dimension of nanometer level satisfying the condition for the ion-selective permeation. Figure 9 shows the fluorescence-time profile detected on the anodic end of the nanochannel, that is, a depletion curve. Compared to the fluorescence intensities of the FITC dye of various concentration filled on the microchannel without bias voltage applied, a depletion factor of 200 was estimated, indicating the effectiveness of the hybrid micro- and nano-fluidic PMMA chip for ion removing.

The fluorescent images taken during ion enrichment–depletion of FITC dye ions on a fluidic chip with hybrid micro- and nano-channels. a A schematic diagram for the fluidic chip. The two U-shaped microchannels were 2 cm in length, 100 μm in top width, 200 μm in bottom width, and 100 μm in depth. The nanogap connecting the microchannels was 1 mm in length, 90 μm in width, and 100 nm in lithographied depth. b The channels were filled with a solution of 20 μmol l−1 FITC in 50 μmol l−1 borate buffer (pH 8.2), and no bias voltage was applied. c and d 3 s and 18 s after a voltage of 1.0 kV was applied to the microchannels, respectively. The symbols of “−” and “+” represent the cathode end and anode end of the nanogap, respectively

The depletion curve of the FITC dye ions collected at the anode end of the nanochannel during the ion enrichment–depletion process
4 Conclusion
A novel method for fabrication of 1-D nanochannels or hybrid micro- and nano-channel network on PMMA sheets was reported here. The nanochannels with specified depth could be resist-free UV-lithographied on the PMMA substrate with regulation of the UV dose exposed to the PMMA surface. Secondary UV-treatment of the surfaces of the PMMA substrates with the channel structure and the flat PMMA cover sheets is necessary for the low-temperature and low-pressure bonding of the chips. The developed method features simple in operation, low in fabrication cost, needless of a clean room, and expensive equipments. With the developed method, chemists or biologists can fabricate PMMA fluidic chips with nanochannels or hybrid micro- nano-channel network in common wet laboratories. With simple modifications, this method is potential to be used for fabrication of nanofluidic chips made of such polymers such as PC, poly(ethylene terephthalate), polystyrene chips, etc.
References
Abgrall P, Nguyen NT (2008) Nanofluidic devices and their applications. Anal Chem 80:2326–2341
Abgrall P, Low LN, Nguyen NT (2007) Fabrication of planar nanofluidic channels in a thermoplastic by hot-embossing and thermal bonding. Lab Chip 7:520–522
Cabodi M, Turner SWP, Craighead HG (2002) Entropic recoil separation of long DNA molecules. Anal Chem 74:5169–5174
Cannon DM, Flachsbart BR, Shannon MA, Sweedler JV, Bohn PW (2004) Fabrication of single nanofluidic channels in poly(methylmethacrylate) films via focused-ion beam milling for use as molecular gates. Appl Phys Lett 85:1241–1243
Chantiwas R, Hupert ML, Pullagurla SR, Balamurugan S, López JT, Park S, Datta P, Goettert J, Cho YK, Soper SA (2010) Simple replication methods for producing 1D nanoslits in thermoplastics and the transport dynamics of double-stranded DNA through these slits. Lab Chip 10:3255–3264
Cho YH, Park J, Park H, Cheng X, Kim BJ, Han A (2010) Fabrication of high-aspect-ratio polymer nanochannels using a novel Si nanoimprint mold and solvent-assisted sealing. Microfluid Nanofluid 9:163–170
Gardeniers HJGE (2009) Chemistry in nanochannel confinement. Anal Bioanal Chem 394:385–397
Gates BD, Xu QB, Stewart M, Ryan D, Willson CG, Whitesides GM (2005) New approaches to nanofabrication: molding, printing, and other techniques. Chem Rev 105:1171–1196
Ginger DS, Zhang H, Mirkin CA (2004) The evolution of dip-pen nanolithography. Angew Chem Int Ed 43:30–45
Haiducu M, Rahbar M, Foulds IG, Johnstone RW, Sameoto D, Parameswaran M (2008) Deep-UV patterning of commercial grade PMMA for low-cost, large-scale microfluidics. J Micromech Microeng 18:115029
Han J, Craighead HG (2000) Separation of long DNA molecules in a microfabricated entropic trap array. Science 288:1026–1029
He QH, Chen S (2008) Fabricating technologies of nanofluidic chips and their applications. Prog Chem (Chin J) 12:2061–2067
He QH, Chen S, Su Y, Fang Q, Chen HW (2008) Fabrication of 1D nanofluidic channels on glass substrate by wet etching and room-temperature bonding. Anal Chim Acta 628:1–8
Huang XH, Gordon MJ, Zare RN (1988) Current-monitoring method for measuring the electroosmotic flow rate in capillary zone electrophoresis. Anal Chem 60:1837–1838
Jia ZJ, Fang Q, Fang ZL (2004) Bonding of glass microfluidic chips at room temperatures. Anal Chem 76:5597–5602
Johnstone RW, Foulds IG, Parameswaran M (2008) Deep-UV exposure of poly(methyl methacrylate) at 254 nm using low-pressure mercury vapor lamps. J Vac Sci Technol B 26:682–685
Kaczmarek H, Kaminska A, van Herk A (2000) Photooxidative degradation of poly(alkyl methacrylate)s. Eur Polym J 36:767–777
Kim SM, Burns MA, Hasselbrink EF (2006) Electrokinetic protein preconcentration using a simple glass/poly(dimethylsiloxane) microfluidic chip. Anal Chem 78:4779–4785
Kim SJ, Song YA, Han JY (2010) Nanofluidic concentration devices for biomolecules utilizing ion concentration polarization: theory, fabrication, and applications. Chem Soc Rev 39:912–922
Kong Y, Chen HW, Wang YR, Steven SA (2006) Fabrication of a gold microelectrode for amperometric detection on a polycarbonate electrophoresis chip by photodirected electroless plating. Electrophoresis 27:2940–2950
Kong Y, Chen HW, Yun X, Hao ZX, Fang ZL (2007) A simple photomask with photoresist mask layer for ultraviolet-photolithography and its application for selectively photochemical surface modification of polymers. Chin J Anal Chem 35:623–627
Kovarik ML, Jacobson SC (2009) Nanofluidics in lab-on-a-chip devices. Anal Chem 81:7133–7140
Krishnan M, Monch I, Schwille P (2007) Spontaneous stretching of DNA in a two-dimensional nanoslit. Nano Lett 7:1270–1275
Lee JH, Chung S, Kim SJ, Han JY (2007) Poly(dimethylsiloxane)-based protein preconcentration using a nanogap generated by junction gap breakdown. Anal Chem 79:6868–6873
Lei KF, Ahsan S, Budraa N, Li WJ, Mai JD (2004) Microwave bonding of polymer-based substrates for potential encapsulated micro/nanofluidic device fabrication. Sens Actuator A 114:340–346
Lippert T, Dickinson JT (2003) Chemical and spectroscopic aspects of polymer ablation: special features and novel directions. Chem Rev 103:453–485
Mahabadi KA, Rodriguez I, Haur SC, van Kan JA, Bettiol AA, Watt F (2006) Fabrication of PMMA micro- and nanofluidic channels by proton beam writing: electrokinetic and morphological characterization. J Micromech Microeng 16:1170–1180
Mao P, Han JY (2005) Fabrication and characterization of 20 nm planar nanofluidic channels by glass–glass and glass–silicon bonding. Lab Chip 5:837–844
McCarley RL, Vaidya B, Wei SY, Smith AF, Patel AB, Feng J, Murphy MC, Soper SA (2005) Resist-free patterning of surface architectures in polymer-based microanalytical devices. J Am Chem Soc 127:842–843
Mijatovic D, Eijkel JCT, van den Berg A (2005) Technologies for nanofluidic systems: top-down vs. bottom-up—a review. Lab Chip 5:492–500
Napoli M, Eijkel JCT, Pennathur S (2010) Nanofluidic technology for biomolecule applications: a critical review. Lab Chip 10:957–985
O’Brien MJ, Bisong P, Ista LK, Rabinovich EM, Garcial AL, Sibbett SS, Lopez GP, Brueck SRJ (2003) Fabrication of an integrated nanofluidic chip using interometric lithography. J Vac Sci Technol B 21:2941–2945
Perry JL, Kandlikar SG (2006) Review of fabrication of nanochannels for single phase liquid flow. Microfluid Nanofluid 2:185–193
Pu QS, Yun J, Temkin H, Liu SR (2004) Ion-enrichment and ion-depletion effect of nanochannel structures. Nano Lett 4:1099–1103
Sivanesan P, Okamoto K, English D, Lee CS, DeVoe DL (2005) Polymer nanochannels fabricated by thermomechanical deformation for single-molecule analysis. Anal Chem 77:2252–2258
Srinivasan R, Lazare S (1985) Modification of polymer surfaces by far-ultraviolet radiation of low and high (laser) intensities. Polymer 26:1297–1300
Tsao CW, DeVoe DL (2009) Bonding of thermoplastic polymer microfluidics. Microfluid Nanofluid 6:1–16
Tsao CW, Hromada L, Liu J, Kumar P, Devoe DL (2007) Low temperature bonding of PMMA and COC microfluidic substrates using UV/ozone surface treatment. Lab Chip 7:499–505
Wang YC, Stevens AL, Han JY (2005) Million-fold preconcentration of proteins and peptides by nanofluidic filter. Anal Chem 77:4293–4299
Wang XY, Wang SL, Veerappan V, Byun CK, Nguyen H, Gendhar B, Allen RD, Liu SR (2008) Bare nanocapillary for DNA separation and genotyping analysis in gel-free solutions without application of external electric field. Anal Chem 80:5583–5589
Wei SY, Vaidya B, Patel AB, Soper SA, McCarley RL (2005) Photochemically patterned poly(methyl methacrylate) surfaces used in the fabrication of microanalytical devices. J Phys Chem B 109:16988–16996
Wu DP, Steckl AJ (2009) High speed nanofluidic protein accumulator. Lab Chip 9:1890–1896
Yang B, Dukkipati VR, Li D, Cardozo BL, Pang SW (2007) Stretching and selective immobilization of DNA in SU-8 micro- and nanochannels. J Vac Sci Technol B 25:2352–2356
Yasin S, Hasko DG, Ahmed H (2001) Fabrication of <5 nm width lines in poly(methylmethacrylate) resist using a water: isopropyl alcohol developer and ultrasonically-assisted development. Appl Phys Lett 78:2760–2762
Yu H, Lu Y, Zhou YG, Wang FB, He FY, Xia XH (2008) A simple, disposable microfluidic device for rapid protein concentration and purification via direct-printing. Lab Chip 8:1496–1501
Zhang JY, Esrom H, Kogelschatz U, Emig G (1994) Modification of polymers with UV excimer radiation from lasers and lamps. J Adhes Sci Technol 8:1179–1210
Acknowledgments
This study is funded by the National Natural Science Foundation of China (project No. 20775068 and 20890020) and the National Basic Research Program of China (973 Program, project No. 2007CB714502). Professor Qun Fang is thanked for valuable discussion.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Hu, X., He, Q., Zhang, X. et al. Fabrication of fluidic chips with 1-D nanochannels on PMMA substrates by photoresist-free UV-lithography and UV-assisted low-temperature bonding. Microfluid Nanofluid 10, 1223–1232 (2011). https://doi.org/10.1007/s10404-010-0753-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10404-010-0753-6