
Abstract
Performing medical diagnosis in microfluidic devices could scale down laboratory functions and reduce the cost for accessible healthcare. The ultimate goal of such devices is to receive a sample of blood, perform genetic amplification (polymerase chain reaction—PCR) and subsequently analyse the amplified products. DNA amplification is generally performed with DNA purified from blood, thus requiring on-chip implementation of DNA extraction steps with consequent increases in the complexity and cost of chip fabrication. Here, we demonstrate the use of unprocessed whole blood as a source of template for genomic or viral targets (human platelet antigen 1 (HPA1), fibroblast growth factor receptor 2 (FGFR2) and BK virus (BKV)) amplified by PCR on a three-layer microfluidic chip that uses a flexible membrane for pumping and valving. The method depends upon the use of a modified DNA polymerase (Phusion™). The volume of the whole blood used in microchip PCR chamber is 30 nl containing less than 1 ng of genomic DNA. For BKV on-chip whole blood PCR, about 3000 copies of BKV DNA were present in the chamber. The DNA detection method, laser-induced fluorescence, used in this article so far is not quantitative but rather qualitative providing a yes/no answer. The ability to perform clinical testing using whole blood, thereby eliminating the need for DNA extraction or sample preparation prior to PCR, will facilitate the development of microfluidic devices for inexpensive and faster clinical diagnostics.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
PCR, one of the most sensitive techniques for detecting pathogens or genes, is frequently used by clinical and research laboratories for medical testing. In many cases the pathogens or clinically relevant biomarkers are found in the peripheral circulation, which means that the relevant nucleic acid templates must be detected in blood (Eisenstein 1990). Usually, DNA must be purified to remove blood components that inhibit PCR (Higuchi 1989; Akane et al. 1994). Since DNA extraction procedures involve time-consuming steps and require skilled technicians, the use of unprocessed whole blood would be advantageous, particularly for miniaturised technologies seeking to automate PCR. Reports of PCR with whole blood involve a buffer with higher pH (Bu et al. 2008), a novel reagent cocktail (Ampdirect buffer) (Nishimura et al. 2000), different Taq DNA polymerases (Kermekchiev et al. 2009), or heating of diluted blood for 15 min before PCR (McCusker et al. 1992), all using conventional thermocycling. In theory, PCR on microfluidic chips offers multiple advantages over conventional methods, including faster speed, smaller samples, less reagent usage, with integration and automation of the entire process, from introduction of unprocessed sample to detection of PCR product (Chen et al. 2007). Despite these advances in developing microfluidic devices, most on-chip PCR has been performed with plasmids or purified DNA as templates (Liu et al. 2006; Wang and Burns 2009). We previously performed on-chip PCR to amplify BKV directly from raw urine (Kaigala et al. 2006). House et al. (2010) used unpurified methicillin-resistant Staphylococcus aureus (MRSA) DNA for real-time PCR on a microfluidic chip. Several groups have developed on-chip sample purification (SP) techniques for extracting DNA from whole blood prior to on-chip PCR (Easley et al. 2006; Chia et al. 2010; Lien et al. 2009; Price et al. 2009). However, implementing SP on-chip complicates fabrication and instrumentation needed for SP/PCR. In this article, we report a method to directly amplify viral or genomic DNA templates from whole blood using a three-layer chip with a PDMS membrane for pumping/valving, and an inexpensive prototype instrument to amplify two genomic DNA targets and one viral DNA target in nanolitre volumes. Whole blood was added to a microfluidic chip, followed by amplification of HPA1 or FGFR2, and of a template from BKV. To our knowledge, this is the first report of on-chip PCR without DNA purification, using whole blood as a source of template.
2 Experimental procedure
2.1 Microfluidic chips
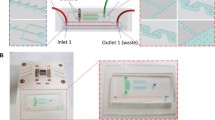
The microfluidic chip used for PCR and CE consists of a flexible polydimethylsiloxane (PDMS—Sylgard 184, Dow Corning) layer sandwiched between two layers of Borofloat glass substrates (Schott AG, Germany, 1.1 mm thickness) that are etched with wells and channels for the reaction and for PDMS-mediated pumping/valving (Fig. 1a, b). The glass microchips were fabricated by standard glass etching processes as previously described in detail (Kaigala et al. 2006). The microfluidic channels in the top glass layer are 45 μm deep while PCR chamber is 90 μm deep. The channels required for controlling the valves in the bottom glass layer are 70 μm deep. PDMS membranes were fabricated with the monomer and curing agent mixed in a 10:1 weight ratio to a thickness of about 300 μm. PDMS membrane was irreversibly bonded to the upper and lower etched glass plates by exposing the surfaces of PDMS and glass to UV light for 7 min. The pumping and valving were done by Mathies-style valves (Grover et al. 2003) where three valves can be used as a pump by actuating them in an appropriate sequence in order to pump the reagents from PCR regents loading well to the PCR chamber and from PCR chamber to the sample reservoir post-PCR. The volume of the PCR chamber is 600 nl.

a Schematic of the PCR/CE glass/PDMS/glass microfluidic chip and b photograph of the chip. Chip dimensions are 95 × 18 mm2. The volume of the PCR chamber is 600 nl. Heights of the chamber and the channels are 90 and 45 μm, respectively. The six ports on the right of the PCR chamber and valves in the photograph are for connection of vacuum and pressure lines for pumping fluid and sealing the chamber
2.2 Instrumentation
An inexpensive prototype instrument was used to perform on-chip PCR reactions (Fig. 2a, b). It uses a Motorola 68332 microprocessor to control a Peltier element (XLT2398-01L, Marlow Industries, Dallas, TX) for heating and cooling during PCR. The system is calibrated by placing a calibrated thermocouple (5TC-TT-K-40-36, Omega Engineering Inc., Stamford, CT) in the PCR chamber of a similar chip. Prior to the PCR, parameters such as the PCR cycle times, temperatures, and number of cycles are programmed by the user into the microcontroller via the LCD screen. For actuating pumps for fluidic handling, pressure and vacuum were generated by mini diaphragm pumps (P/N VMP1624MM-12-90, Virtual Industries, Inc., Colorado Springs, CO) that are controlled by the microcontroller.

a Schematic representation showing major components and b photograph of the prototype instrument used for PCR. The microfluidic chip is placed on the peltier element in the drawer as shown in b. The drawer was then closed and the gantry was lowered by turning the knob on the top of the instrument. This caused a seal for pressure and vacuum lines to facilitate fluid movement within the chip. Lowering the gantry also lowered Thermocouple 2 so that the temperature readings could be taken on the top surface of the chip
2.3 Whole blood amplification
Whole blood amplification of target regions was performed on-chip on HPA1 or FGFR2, and for BKV. As positive controls, purified DNA was amplified on-chip for HPA1 and BKV, or on a conventional thermocycler for FGFR2. For on-chip whole blood PCR, the buffer and Taq polymerase were from Phusion® Blood Direct PCR Kit (Cat. #F-547, Finnzymes, Espoo, Finland). Blood was obtained from healthy donors as approved by the University of Alberta Health Ethics Review Board, and stored at −30°C. The details of each PCR are shown below.
Prior to PCR with purified DNA as template, the channels and PCR chamber were incubated in 1% BSA (bovine serum albumin, Sigma, St. Louis, MO) for 1 h. For PCR utilising whole blood as template, no BSA coating was performed. We found that the performance of the whole blood PCR is not affected by the BSA coating, in contrast to our observations that BSA coating is essential for amplification of templates in purified DNA. The protein components in whole blood may be coating the surface of PDMS, obviating the need for surface pre-treatment prior to PCR. In order to perform PCR, 5 μl of PCR mix was added to the PCR loading well and pumped into the PCR chamber by actuating valves. Reaction mixes and the cycle conditions for each PCR are listed below.
2.3.1 HPA1 amplification from whole blood
Primers for the HPA1 PCR were 5′-ATAGCTCTGATTGCTGGACTTC-3′ (Forward, 10 μM) and Cy5-5′-GATTCTGGGGCACAGTTATCC-3′ (Reverse, 10 μM, Cy5 labelled). For HPA1 whole blood PCR, the 20 μl reaction mix consisted of 10 μl of 2× Phusion buffer, 1 μl of each primer, 0.4 μl of Phusion Blood DNA polymerase, 1 μl of whole blood and PCR grade water (Fluka Analytical, Buchs SG, Switzerland). For HPA1 DNA PCR, 20 μl reaction mix consisted of 2 μl of 10× PCR buffer (Invitrogen, Carlsbad, CA), 0.4 μl of each primer, 0.4 μl of 10 mM dNTP (Invitrogen), 1.4 μl of 50 mM MgCl2 (Invitrogen), 0.4 μl of 1% BSA, 2U Platinum® Taq polymerase (Invitrogen), 50 ng of purified genomic DNA and water. PCR was performed with an initial 94°C—120 s denaturation step followed by denaturation, annealing and extension steps of 94°C for 30 s, 59°C for 30 s and 70°C for 20 s, respectively, for 35 cycles, and a final extension step of 72°C for 120 s. 10 μl of each reaction mix was PCR cycled in the thermocycler as controls.
2.3.2 BKV amplification from whole blood
Primers for the BKV PCR were Alexa-5′-GTGACCAACACAGCTACCACAGTGT-3′ (Forward, 10 μM, Alexa labelled) and Alexa-5′-TCAAACACCCTAACCTCTTCTACCTG-3′ (Reverse, 10 μM, Alexa labelled). For BKV whole blood PCR, the reaction mix was similar to the HPA1 whole blood PCR described above. To mimic BK viremia, 1 μl of whole blood was spiked with 105 copies of purified BKV DNA before adding to the BKV PCR mix. For BKV purified DNA PCR, 20 μl reaction mix consisted of 2 μl of 10× PCR buffer, 0.4 μl of each primer, 0.4 μl of 10 mM DNTP, 0.5 μl of 1% BSA, 2U Platinum® Taq polymerase, 1.6 μl of 50 mM MgCl2, 0.4 μl of BKV template DNA having a titre of 108 copies/ml, 0.8 μl of DMSO (dimethylsulphoxide, Fisher, Fair Lawn, NJ) and water. A short two-step PCR was performed for BKV with an initial 94°C—120 s denaturation step followed by 94°C for 10 s and 60°C for 20 s for 35 cycles, and a final extension step of 72°C—60 s. Total PCR time was about 40 min.
2.3.3 FGFR2 amplification from whole blood
For the FGFR2 PCR, Phusion® reaction mix was as described for the HPA1 PCR reaction except for the primers. Primers for FGFR2 PCR were 5′-CAGAAGTTTTTGAGAGTGGCATGATG (Forward, 10 μM) and 5′-GCTGACTTCTATTTATATAACTTCAAGC (Reverse, 10 μM). On-chip PCR was performed with an initial 94°C—120 s denaturation step followed by 94°C for 10 s, 64°C for 20 s and 72°C for 20 s for 35 cycles, and a final extension step of 72°C—120 s. Gradient PCR was performed at thermocycler with similar PCR parameters to the chip but with different annealing temperatures.
2.4 Detection of amplicons using capillary electrophoresis
Detection of HPA1 and BKV PCR products were performed by microchip capillary electrophoresis (CE) on a Microfluidic Tool Kit (Micralyne, Edmonton, AB) that includes high-voltage electronics for DNA manipulation through the channels, optics and optical detection electronics for detection of DNA by laser-induced fluorescence (LIF) and software (Manage et al. 2005). The excitation wavelength of the laser is 635 nm while the detection is done at 670 nm. A compiled LabVIEW interface supplied with the Microfluidc Tool Kit was used to record the LIF signal at 200 Hz. The sieving matrix used for DNA separation was 4% linear polyacrylamide (LPA, cat# 19901, Polysciences, Inc, Warington, PA) in 1× Tris TAPS EDTA (TTE) buffer. The sample waste, buffer reservoir and buffer waste wells were each filled with 4 μl of 1× TTE buffer. When the PCR was completed, the contents in the PCR chamber were flushed out to the sample reservoir by loading 5 μl of 0.1× TTE buffer into the loading well and pumping it through the PCR well. In the Microfluidic Tool Kit, PCR product was injected from the sample reservoir towards the sample waste well by applying 300 V across them (ca. 333 V/cm) for 90 s. The DNA was then separated along the separation channel by applying 600 V through the buffer reservoir and the buffer waste (ca. 67 V/cm) for 180 s. The detection was done at 12 mm along the separation channel.
3 Results and discussion
3.1 Amplification of HPA1
The electropherograms of HPA1 on-chip PCR products with whole blood as well as with purified DNA are shown in Fig. 3. The length of the HPA1 PCR product is 115 bp. The electropherogram of the thermocycler PCR performed with whole blood is also shown. CE of the size standard (ALFfexpress, 50–500 bp, Amersham Biosciences, NJ, USA) confirms the size of the PCR product. The use of Phusion® Blood Direct PCR Kit for purified DNA amplification results in non-specific peaks when a reaction mix similar to the blood-PCR is used.

Electropherograms showing on-chip HPA1 PCR (115 bp) products amplified from purified DNA and from whole blood on-chip and on the thermocycler (TC), and the ALFexpress size standard. The first peak in the electropherograms is due to primer dimers. Electrophoregrams are offset along the Y-axis for clarity
3.2 Amplification of BKV
Figure 4 shows the electropherograms of on-chip BKV PCR (295 bp) products performed with whole blood and purified DNA, and the electropherogram of the thermocycler PCR performed with whole blood. The sizes of both HPA1 and BKV PCR products were also confirmed with conventional gel electrophoresis (not shown). The large primer dimer peak in BKV on-chip PCR in Fig. 4 is likely to be due to the overloading of the primer dimers, causing them to appear later than expected (ABI 2009). Representative PCR products were sequenced with ABI 3130xl DNA capillary analysis system (Applied Biosystems, Foster City, CA), as described previously (Adamia et al. 2008), by pooling PCR products from multiple on-chip PCRs.

Electropherograms showing on-chip BKV PCR (295 bp) products amplified from purified DNA and from whole blood, thermocycler (TC) product amplified from the whole blood, and the ALFexpress size standard. The first peak in the electropherograms is due to primer dimers. Electrophoregrams are offset along the Y-axis for clarity
3.3 Amplification of FGFR2
FGFR2 amplicons (272 bp) from on-chip PCR and thermocycler PCR are shown in Fig. 5. The volume of the conventional PCR used for the gel electrophoresis of Fig. 5 was 10 μl, while the volume in the on-chip PCR was 600 nl. Therefore, the amount of whole blood used in conventional PCR is 0.5 μl per reaction, ~17 times more than for the on-chip PCR which used 30 nl of whole blood per reaction. This explains the difference between the intensities of the thermocycler PCR products and the on-chip PCR product. Representative PCR products were verified by sequencing.

Whole blood PCR on FGFR2 gene (272 bp): off-chip and on-chip. Lanes 1–4 gradient PCR performed off-chip on the thermocycler with annealing temperatures from 60.8, 62.0, 63.4 and 65.9°C, respectively (10 μl each), Lane 5 DNA ladder (exACTGene™; Low Range Plus DNA Ladder, Fisher), Lane 6 not loaded-empty and Lane 7 on-chip PCR (~600 nl)
3.4 Advantages for the use of whole blood PCR on chip
Using whole blood as the source of template, we have successfully demonstrated PCR reactions on microfluidic chips for three different DNA templates, using nanolitre reaction volumes. The use of whole blood avoids tedious and laborious DNA isolation processes that restrict the development and automation of fast diagnostic procedures. Whole blood PCR depends on the use of a polymerase that is not inhibited by components of whole blood, in this case a commercially available enzyme termed Phusion®. Phusion polymerase, a hot start polymerase, uses a reversibly binding Affibody® protein (Nord et al. 1997). With the Phusion® DNA polymerase, blood volumes in the PCR reaction mix can be up to 40%. We used a concentration of 5% of whole blood for on chip or conventional PCR reactions (1 μl whole blood in 20 μl reaction mix).
4 Conclusion
This work shows the successful use of whole blood as a source of template for amplification on chip of genomic DNA (HPA1 and FGFR2) and BK virus. The ability to perform amplification of templates in whole blood using on-chip PCR eliminates the need for DNA extraction and purification procedures hence simplifying the development and fabrication of integrated microfluidic devices and instruments to perform affordable medical diagnostics.
References
ABI (2009) DNA sequencing by capillary electrophoresis. Applied Biosystems Chemistry Guide
Adamia S, Reichert AA, Kuppusamy H, Kriangkum J, Ghosh A, Hodges JJ, Pilarski PM, Treon SP, Mant MJ, Reiman T, Belch AR, Pilarski LM (2008) Inherited and acquired variations in the hyaluronan synthase 1 (HAS1) gene may contribute to disease progression in multiple myeloma and Waldenstrom macroglobulinemia. Blood 112:5111–5121
Akane A, Matsubara K, Nakamura H, Takahashi S, Kimura K (1994) Identification of the heme compound copurified with deoxyribonucleic-acid (DNA) from bloodstains, a major inhibitor of polymerase chain-reaction (PCR) amplification. J Forensic Sci 39:362–372
Bu Y, Huang H, Zhou G (2008) Direct polymerase chain reaction (PCR) from human whole blood and filter-paper-dried blood by using a PCR buffer with a higher pH. Anal Biochem 375:370–372
Chen L, Manz A, Day PJR (2007) Total nucleic acid analysis integrated on microfluidic devices. Lab Chip 7:1413–1423
Chia BT, Yang XY, Cheng MY, Yang YJ (2010) A DNA extraction and polymerase-chain-reaction microchip using magnetic beads and thermo-pneumatic valves. Mems 2010: 23rd IEEE international conference on micro electro mechanical systems, Technical Digest, pp 967–970
Easley CJ, Karlinsey JM, Bienvenue JM, Legendre LA, Roper MG, Feldman SH, Hughes MA, Hewlett EL, Merkel TJ, Ferrance JP, Landers JP (2006) A fully integrated microfluidic genetic analysis system with sample-in-answer-out capability. Proc Natl Acad Sci USA 103:19272–19277
Eisenstein BI (1990) The polymerase chain-reaction—a new method of using molecular-genetics for medical diagnosis. N Engl J Med 322:178–183
Grover WH, Skelley AM, Liu CN, Lagally ET, Mathies RA (2003) Monolithic membrane valves and diaphragm pumps for practical large-scale integration into glass microfluidic devices. Sens Actuators B Chem 89:315–323
Higuchi R (1989) PCR technology: principles and applications for DNA amplification. Stockton Press, New York
House DL, Chon CH, Creech CB, Skaar EP, Li DQ (2010) Miniature on-chip detection of unpurified methicillin-resistant Staphylococcus aureus (MRSA) DNA using real-time PCR. J Biotechnol 146:93–99
Kaigala GV, Huskins RJ, Preiksaitis J, Pang XL, Pilarski LM, Backhouse CJ (2006) Automated screening using microfluidic chip-based PCR and product detection to assess risk of BK virus-associated nephropathy in renal transplant recipients. Electrophoresis 27:3753–3763
Kermekchiev MB, Kirilova LI, Vail EE, Barnes WM (2009) Mutants of Taq DNA polymerase resistant to PCR inhibitors allow DNA amplification from whole blood and crude soil samples. Nucleic Acids Res 37:e40
Lien KY, Liu CJ, Lin YC, Kuo PL, Lee GB (2009) Extraction of genomic DNA and detection of single nucleotide polymorphism genotyping utilizing an integrated magnetic bead-based microfluidic platform. Microfluid Nanofluidics 6:539–555
Liu CN, Toriello NM, Mathies RA (2006) Multichannel PCR-CE microdevice for genetic analysis. Anal Chem 78:5474–5479
Manage DP, Zheng Y, Somerville MJ, Backhouse CJ (2005) On-chip HA/SSCP for the detection of hereditary haemochromatosis. Microfluid Nanofluidics 1:364–372
McCusker J, Dawson MT, Noone D, Gannon F, Smith T (1992) Improved method for direct PCR amplification from whole-blood. Nucleic Acids Res 20:6747
Nishimura N, Nakayama T, Tonoike H, Kojima K, Kato S (2000) Direct polymerase chain reaction from whole blood without DNA isolation. Ann Clin Biochem 37:674–680
Nord K, Gunneriusson E, Ringdahl J, Stahl S, Uhlen M, Nygren PA (1997) Binding proteins selected from combinatorial libraries of an alpha-helical bacterial receptor domain. Nat Biotechnol 15:772–777
Price CW, Leslie DC, Landers JP (2009) Nucleic acid extraction techniques and application to the microchip. Lab Chip 9:2484–2494
Wang F, Burns MA (2009) Performance of nanoliter-sized droplet-based microfluidic PCR. Biomed Microdevices 11:1071–1080
Acknowledgments
This work was supported by an Interdisciplinary Team Grant from the Alberta Heritage Foundation for Medical Research (AHFMR). LMP is the Canada Research Chair in Biomedical Nanotechnology and this work was funded in part by the Chair’s program. We thank Dr. Xiao-Li Pang, Provincial Laboratories for Public Health, Edmonton, Alberta for providing BKV DNA and Paul R. Dumais for the fabrication of the chips.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Manage, D.P., Morrissey, Y.C., Stickel, A.J. et al. On-chip PCR amplification of genomic and viral templates in unprocessed whole blood. Microfluid Nanofluid 10, 697–702 (2011). https://doi.org/10.1007/s10404-010-0702-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10404-010-0702-4