
Abstract
Robust, high temperature micro-reactors for on-line conversion of organic compounds were microfabricated in high purity fused silica to enable stable isotopic compositional analysis of individual compounds in mixtures using advanced gas chromatography (GC) separation techniques, such as fast GC and comprehensive 2D GC, coupled to isotope ratio mass spectrometry (IRMS). These micro-reactors could also be manufactured at larger channel dimensions to enable robust operations for normal GCC-IRMS applications. Photolithography was used to define the reactor channel pattern on high purity fused silica, with a protective layer of amorphous silicon. A two-step isotropic wet etching process, using 49% HF, selectively created semi-circular cross-section micro-channels of arbitrary diameters (56–209 μm), with tapered sections leading to input/output ports (>400 μm) that accept fused silica capillary tubing used in GC and MS peripherals. Pairs of symmetric mirror image substrates were aligned and bonded to form enclosed circular channels. The resulting micro-reactors are more robust than the standard designs made of fragile-fused silica capillary or alumina tubes of relatively large bore, and are gas tight at temperatures up to 1,000°C. Fast GC plugs of CH4 with and without the reactor revealed that peak shapes are minimally affected by the micro-reactor when carrier flow rate and channel diameter are optimized. Peak shapes with full widths at half maximum of 250 ms are shown for plugs of CH4 through a fast-GC-combustion-IRMS system interfaced with a micro-reactor containing a CuO/NiO combustion source, enabling carbon isotope ratio measurements of CH4 with a precision of SD(δ13C) = ±0.28‰. The devices enable arbitrarily narrow bores and long path channels that can be operated as open tubes or loaded with a reactant, in a small, robust package.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
High precision gas isotope ratio mass spectrometry (IRMS) is the established technique for the measurement of the natural isotopic variability of the organic elements, C, N, O, S, and H, which enables the determination of the geographic, chemical, and biological origins of substances, and is employed in a wide range of disciplines. Some examples include detection of athletic steroid doping, the sourcing and authentication of foods, natural products, and drugs, non-radioactive tracer analysis for biomedical research, and a plethora of forensics applications. In carbon isotope ratio analysis of 13C/12C, for example, the analyte must be converted to CO2 for analysis; therefore, most analytes must be chemically transformed by combustion (Asche et al. 2003; Brenna et al. 1997; Sessions 2006). Compound-specific isotope analysis was enabled by the integration of gas chromatography (GC) with IRMS via an online combustion interface (GCC-IRMS) (Matthews and Hayes 1978). In other schemes, decomposition products of an individual compound by pyrolysis at high temperatures are of interest, and the products are either measured by molecular mass spectrometry and/or chemically converted in a second reactor in order to measure their isotopic composition by IRMS (Corso and Brenna 1997).
The reactors employed in these systems are normally embodied as an alumina (ceramic) tube (Brand 1995; Hayes 1991; Matthews and Hayes 1978) loaded with a reactant of interest, or no reactant and/or a catalyst in the case of pyrolysis (Burgoyne and Hayes 1998; Corso and Brenna 1997; Tobias and Brenna 1997). In order to improve peak shapes for chromatography by reducing dead volumes associated with connections, a continuous fused silica capillary design was reported and refined (Corso and Brenna 1997; Ellis and Fincannon 1997; Goodman 1995; Goodman 1998; Sacks et al. 2007; Tobias et al. 2008) and is in use in several laboratories. Apart from the input and output ends, the reactor is held in a high-temperature furnace. For example, in 13C/12C analysis, CuO, heated between 850 and 960°C, is used as a source of O2 in order to combust organic molecules to CO2 gas. The dimensions of the reactors range from inner diameters (i.d.) of 500 μm for the alumina tubes, down to the state of the art of 250 μm i.d. for fused silica capillary. The lengths of the reaction hot zone in currently employed systems range from 150 to 200 mm.
Recently, the coupling of these systems to more advanced upstream GC separation techniques have been demonstrated by our laboratory, using a 250 μm i.d. GC fused silica capillary reactor, to enable more rapid separation by fast-GCC-IRMS (Sacks et al. 2007) and improved separations of complex sample mixtures by comprehensive 2D GC (GCxGCC-IRMS) (Tobias et al. 2008). These techniques require the preservation of very narrow peak shapes produced in the gas-phase separations; therefore, post-column band broadening can be further minimized by reducing the i.d. of the reactor from the current value of 250 μm. In addition, improvement of the physical stability of the reactor is required at temperatures up to 1,000°C and beyond. These issues were addressed here, for the first time, by microfabrication to create a micro-reactor. We describe our initial approach towards a microfabricated micro-reactor (MFMR) and its capabilities.
2 Materials and methods
2.1 Microfabrication
2.1.1 Photomask generation
The CAD software utility Layout EditorTM (freeware, http://www.layouteditor.net) was used to create a drawing and an exported graphic data system (GDSII) file of the micro-reactor channel design. The file was used to guide the exposure of photoresist on a chromium coated, 5 inch (124 mm) square glass substrate using a GCA Mann 3600 Pattern Generator (D. W. Mann/GCA Corporation, USA). The photoresist was developed using 300 MIF developer (Hoechst Celanese, Somerville NJ) for 120 s, water rinsed and spin dried. The exposed chrome layer was removed using CR-14 Chromium Etchant (Cyantek Corporation, Fremont CA) for 120 s, water rinsed and spin dried. Photoresist was subsequently removed by hot stripping in a 75°C bath containing propylene glycol, n-methyl-pyrrolidone (NMP), and tetramethyl ammonium hydroxide (TMAH) for 20 min, water rinsed and spin dried.
2.1.2 Furnace processing
Double side polished 100 mm diameter, 1.0 mm thick, synthetic amorphous high purity fused silica wafers (Corning 7980, Mark Optics, Santa Ana CA) were thoroughly cleaned, as used for metal oxide semiconductor technology, before furnace processing by sequential immersion in a base (6 l H2O, 1 l NH4OH, 1 l H2O2), acid (6 l H2O, 1 l HCl, 1 l H2O2), and HF (20:1 H2O:HF) bath for 10 min, 10 min, and 15 s, respectively. The wafers were water rinsed to 16 MΩ after each bath then spun dry. Low pressure chemical vapor deposition (LPCVD) was used to deposit a layer of approximately 250 nm of amorphous Si using a furnace processing tube (Cryco, Austin TX). Three different Si deposition recipes (SR) were tested. SR1 included a 150 sccm flow of silane (SiH4) at 140 mTorr and 560°C for 100 min. SR2 included a 150 sccm flow of SiH4 at 140 mTorr and 540°C for 250 min. And SR3 included a 150 sccm flow of SiH4 and a 10 sccm flow of 1.5% PH3/N2 (PH3:SiH4 ratio of 1 × 10−3) at 150 mTorr and 540°C for 250 min.
2.1.3 Wafer photolithography
The LPCVD Si surface of each high purity fused silica wafer was dehydrated at 115°C for 1 min on a hot-plate and then cooled. Wafers were then treated and manually spin coated with a solution containing 20% hexamethyldisilazane and 80% propylene glycol monomethyl-ether acetate (MicroPrime P-20 Primer, Shin-Etsi Chemical Company, Japan) as an adhesion promoter, and then spin coated with a 1.3 μm layer of broadband photoresist (Shipley Microdeposit S1813, Rohm & Haas, Philadelphia PA) at a rotational speed of 4,000 rpm for 60 s. Each wafer was then immediately soft-baked at 115°C for 1 min on a hot-plate and then cooled. The photoresist on the wafers was exposed using 405 nm light (20 mW/cm2) for 3.0 s by means of “soft” contact with the photomask using a HTG System III-HR Contact Aligner (Hybrid Technology Group Inc., Scotts Valley CA) The photoresist was developed using 300 MIF developer for 120 s, water rinsed, and spin dried. The exposed amorphous Si surface was plasma etched at 150 W for 11 min using a 30 sccm flow of CF4 at 40 mTorr (Oxford PlasmaLab 80, Oxford Instruments, Oxfordshire, UK).
2.1.4 Isotropic wet etching
The wafers, with the exposed channels and remaining amorphous Si and photoresist, were baked for ~24 h at 90°C before performing a “two step isotropic wet etch”. A solution of 49% HF in water (Mallinckrodt Baker Inc., Phillipsburg NJ) was used as the high purity fused silica wet etchant for creation of channels. The etch rate was measured to be ~1 μm/min at 25°C. Both wafers of a pair that were to be ultimately bonded were etched in the same bath. A pair of wafers were first fully immersed in the etchant for 10–70 min (dependent on desired hot zone channel dimensions), then immersed in water for 5 min, and subsequently dried with air. The pair of wafers was then stood on end with each half facing one another on their wafer flat in the etchant, for an additional amount of time to total ~190 min, so that only a few (5–8) mm of the channel “ends” were immersed and etched more than the rest. The wafers were then immersed in water for 10 min, rinsed, and dried with air. Remaining photoresist was removed by the hot stripping method mentioned above, followed by plasma ashing with reactive oxygen using a Gas Sonics Aura 1000 (Novellus Systems Inc., San Jose CA). The native oxide layer was removed by placing the wafers in a 30:1 buffered oxide etch bath (BOE) for 60 s, then the amorphous Si layer was removed by placing the wafers in a 20% KOH bath.
2.1.5 Channel enclosure and device packaging
Surface roughness of the processed wafers before cleaning and bonding was measured to be <8 angstroms (root mean square) by atomic force microscopy (DI Dimension 3100 AFM, Veeco Instruments, Plainview NY). Extensive cleaning procedures were performed to ensure successful bonding. First, the wafers were cleaned according to the acid/base procedure described above, with the replacement of the HF solution immersion with a second base solution immersion. Then, a 10 min Piranha clean was performed using 1:1 96% H2SO4 and 30% H2O2. Alignment of the features on the two mirrored wafers was performed by hand under an ABM contact aligner microscope (ABM Inc., Scotts Valley CA) and gently compressed in the center by hand to form an initial bond. The wafers were then annealed at 1,100°C for 5 h in a furnace processing tube (Cryco, Austin TX). The micro-reactors were cut out of the bonded wafers using a K&S 7100 Dicing Saw (Advanced Dicing Technologies, Horsham PA). Capillary port openings were created by cutting across the widened channel ends. Appropriate metal reactant in the form of wire was inserted into the hot zone channel loop region. A few millimeter of one end of fused silica capillary tubing (0.5 m × 100 μm i.d. and 360 μm o.d.) was coated with Pyro-Putty 950 (Aremco, Valley Cottage NY), inserted into each of the port regions, and further sealed and secured with TorrSeal (Varian Inc., Palo Alto CA). In one case, the exclusive use of polyimide resin was also tested to coat and seal fused silica capillary into the ports.
2.2 Analytical instrumentation
2.2.1 Fast GC-FID and fast GCC-IRMS
An HP6890A GC-FID (Agilent Technologies, Menlo Park, CA) was used for gas plug injections and flame ionization detection (FID). A MAT 252 IRMS (Thermo Finnigan, Bremen, Germany) was used to measure carbon isotope ratios, 13C/12C. The MAT 252 IRMS was run with the variable ion source conductance window open (by three turns). The RC time constants of all three faraday cup detectors were modified to be 30 ms (Tobias et al. 2008), in order to facilitate accurate data acquisition for the narrow peak shapes observed in this study. The MFMR was placed partially inside an open tubular glass fiber furnace (Thermcraft Inc., Winston-Salem NC). In the case of fast GC-FID experiments, a total length of 1.7 m deactivated fused silica capillary with a 100 μm i.d. was used to couple the MFMR with the GC split/splitless inlet and the FID detector. In the case of fast GCC-IRMS experiments, the same 1.7 m length capillary was used to couple the MFMR with the GC inlet and an additional 0.8 m deactivated fused silica capillary with a 100 μm i.d. was used as a transfer capillary between the MFMR and the IRMS open split. A portion of the effluent was sampled into the IRMS by placement of the upstream end of 1 m × 75 μm i.d. IRMS sampling fused silica capillary nominally flush with the end of the transfer capillary within a Press-Tight® (Siltek®, Restek, Bellefonte, Pa) connector, thereby operated as an open split. Immersion of 6 cm of the transfer capillary into a dry ice/acetone water trap (at −78°C) was used to remove water vapor, generated from the combustion process, before reaching the IRMS.
2.2.2 IRMS data acquisition and 13C/12C analysis
A home-built LabView (National Instruments, Austin, TX) based IRMS data acquisition and reduction system, “SAXICAB”, was used to monitor the m/z 44, 45, and 46 signals, for 44CO2, 45CO2, 46CO2, from the MAT 252 IRMS head amplifiers. The faraday cup detector RC time constants were modified for fast GC peak detection. Details can be found elsewhere (Tobias et al. 2008). All data sets were processed for 13C/12C analysis through SAXICAB. The 13C/12C is normally reported in δ notation with respect to an international standard, in units of per mil (‰). For carbon isotopes, the 13C/12C is expressed as δ13CVPDB = (R SPL − R VPDB)/(R VPDB) × 1,000‰, where R SPL is the 13C/12C of the sample and R VPDB is the 13C/12C of the international standard Vienna PeeDee Belemnite. In this study, the first sample injection in a replicate set was used as the reference for the 13C/12C calculation in order to demonstrate reproducibility.
3 Results and discussion
3.1 Microfabrication
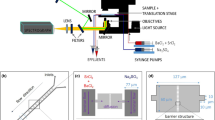
The general MFMR design (Fig. 1a) includes tapered source input and reactor output ports, in addition to a variable hot zone channel region. A photo of a completed MFMR with oxidized metal reactant is shown in Fig. 1b and the test-bed used to characterize the device is shown in Fig. 1c. Reactors were created containing hot zone channel lengths ranging from 150 to 840 mm, channel loop bend radii of 0.35–4.1 mm, and channel diameters varying from 56 to 209 μm. Reference to a specific reactor configuration is compactly made by the code LwwRxxD c yyD w zz, where ww is the Length (L) of the channel in the hot zone in mm, xx is the channel Bend Radius (R) in mm, yy is the hot zone Channel Diameter (D c) in μm before the addition of metal reactant, and zz is the metal reactant Wire Diameter (D w) in μm before oxidation. Photomasks were generated for the micro-reactor patterns with varying hot zone path lengths and channel bend radii (Fig. 2a–f), where white regions are transparent, and made symmetric so that mirror image pairs can be matched later in the process. The hot zone channel region (Fig. 2, i’s) is extended by the input and output ends of the reactor pattern (Fig. 2, ii’s), which become the ports. Both ports lead to the edge of the substrate perpendicular to a wafer flat alignment bar (Fig. 2, iii’s) used in the two step wet etch process described below. The width of the channels in the pattern is made significantly narrower (10 μm) than the ultimately etched channel width, ranging from 56 to 209 μm in this study.

a Schematic of the final version of a micro-reactor (MFMR), before the connection of GC tubing (fused silica capillary) to the source input (GC) and reactor output (FID or IRMS) ports. The hot zone channel region is held at high temperature for combustion. The number, length, and bend radius of the channel loops can be varied as required. Also shown is a b photo of a completed MFMR (L230R3Dc195Dw75) comprised of a 195 μm diameter channel and 75 μm diameter Constantan (55%Cu/45%Ni) wire in the hot zone region of 230 mm in length and 3 mm channel bend radius resulting in an effective diameter of <185 μm. c The same MFMR is shown in a test-bed furnace during operation. MFMR’s were created containing hot zone channels with diameters varying from 56 to 209 μm, hot zone lengths ranging from 150 to 840 mm, and channel bend radii of 0.35–4.1 mm

Example CAD (Layout EditorTM) images of (a–f) photomasks with channel patterns for two micro-reactors each with various (i) hot zone channel lengths and bend radii, including the input and output channel ends (ii), and a wafer flat alignment bar (iii) used in the 2 step wet etch process. Masks are mirror-symmetric to allow enclosure of channels. For convenient reference, photomask patterns are coded LwwRxx, where ww is the Length (L) of the channel in the hot zone in mm, and xx is the channel Bend Radius (R) in mm
High purity fused silica substrates, 100 mm diameter with 1 mm thickness and a wafer flat, were prepared for photolithography (Fig. 3a). After thorough cleaning, one working side was coated with 250 nm of Si as a protective layer mask, which was a coating that is resistant to the HF etchant. The deposition temperature and doping of Si was optimized through investigation of three different low pressure chemical vapor deposition (LPCVD) recipes (SR1, SR2, and SR3). An undoped Si layer formed at higher temperatures (SR1 @ 560°C) formed a layer with excessive defects and pinholes, which appeared to be due to the initiation of crystalline Si formation, and resulted in a poor quality hard mask. A lower LPCVD temperature (SR2 @ 540°C) resulted in a sufficiently amorphous layer of undoped Si with little to no defects, but was difficult to remove later in the process thereby promoting excessive surface roughness. An n-doped amorphous Si layer (SR3 @ 540°C) was ultimately used due to its film quality, resistance to HF, and quick removal using KOH later in the process ensuring clean smooth surfaces for much improved wafer bonding compared to the use of undoped Si. The etch rate of n-doped Si in KOH was approximately 10 times greater than that of undoped Si. After the Si layer was deposited, a broadband positive photoresist was spin coated on top of the hard mask.

Overview of a substrate preparation, b structure fabrication, and c device packaging process work-flows in the microfabrication of the micro-reactor. Depictions of the cross-sections of the substrate, coatings, and etching are shown with first layer on substrate = amorphous Si, second layer on substrate = photoresist, and arrows = 405 nm light
In the structure fabrication process, Fig. 3b, the wafer was aligned under the photomask according to its wafer flat and alignment bar before exposure. The affected photoresist was removed using a developer, defining the channel pattern. These exposed regions in the hard mask were then CF4 plasma etched, thereby uncovering the high purity fused silica surface. A two step isotropic wet etching process was used to create the semi-circular cross-section micro-channels. Each pair of wafers, that were ultimately bonded, was etched in the same bath for optimal uniformity. First, the pair of wafers was fully immersed in 49% HF etchant for a period of time chosen to yield the desired “hot zone” channel dimensions. Second, the pair of wafers was stood on end facing one another on their wafer flat in the etchant, for an additional time period, so that only a few millimeter of the channel ends were immersed and achieved a wider etch diameter than the rest of the pattern. This created a taper from the hot zone channels to the input and output port channels. The HF bath was circulated with a stir bar to ensure uniformity of the etching process over long periods of time. In addition, the bath was physically rotated occasionally to ensure even exposure of non-immersed channels to HF vapors in the fume hood. For example, the reproducibility of the final enclosed hot zone channel dimensions when creating approximately 200 μm diameter channels was 202 ± 4 μm (standard deviation for n = 16 MFMR).
After HF etching, the remaining hard mask was removed by KOH wet etching yielding a smooth, flat substrate surface appropriate for bonding. The surface roughness was measured to be <8 angstroms rms. In the device packaging process (Fig. 3c), the two substrates were aligned under a microscope and pressed together to form the circular channels from the semi-circular cross-sections. Visible fringe lines (i.e., Newton rings) were evidence of gaps between the two wafers. It was imperative that no lines or rings were apparent anywhere near the etched features upon the bonding before the annealing process; otherwise, dead volumes and gas leaks between channels and/or outside of the final device always occurred. The fused silica surfaces adhere by hydrogen bonding attractive forces mediated largely by adsorbed water, maintaining alignment of the two halves. No adhesives are used. The pair was then annealed at high temperature, 1,100°C, for a few hours yielding a permanent gas tight bond due to the creation of Si–O–Si bonds as water is driven away from the interface. The reactor(s) were then diced out of the bonded pair, where capillary port openings were created by cutting across the widened channel ends.
MFMR patterns were varied and tested with hot zone loop bend radii equal to and greater than 3 mm (with patterns L230R3 and L150R4_1). These patterns permit insertion of 75 μm diameter metal reactant wire into channels of the completed device, as is normal for preparation of IRMS combustion reactors. Designs with much tighter loop bend radii can be created; however, deposition of metal reactant prior to bonding may be required. Evaporation, electroplating, and chemical vapor deposition processes are candidate approaches for incorporation into the microfabrication procedure. Fused silica capillary tubing (used for GC) was secured gas tight in each port using Pyro-Putty 950 (maximum operating temperature = 510°C) and TorrSeal (maximum operating temperature = 120°C), and in one case only polyimide resin (maximum operating temperature = 350°C) was evaluated for gas leaks and contained no reactant.
3.2 Physical robustness
MFMR enclosures were successfully constructed consisting of channels with diameters varying from 56 to 209 μm, which are narrower than of previously developed low dead volume fused silica capillary tubing reactors used in IRMS (250–530 μm). A test bench was set up for heat testing of a MFMR at an operating temperature of 950°C (Fig. 1c). A high temperature furnace was resistively heated to 950°C, as measured by a thermocouple, with the serpentine end of a MFMR (L275R0_65Dc85 with no metal reactant) inserted into the hot zone. The inlet capillary was connected to flowing He and the outlet capillary was periodically connected to an IRMS. The MFMR with ports sealed with Pyro-Putty and TorrSeal, was held at 65°C at the port ends and 950°C at the hot zone region representing an 885°C temperature gradient. Tests using Ar gas showed no evidence of leaks over 12 weeks, when the experiment was stopped. Moreover, during other experiments, various MFMR enclosures remained leak free through repeated temperature cycling between room and operating temperature; conventional fragile fused silica capillary reactors frequently crack or shatter due to thermal shock when cycling the reactor between 950°C and room temperature for maintenance or as a result of power interruptions. A preliminary experiment with MFMR ports sealed with polyimide resin resulted in a device similarly resilient to changes in temperature. These observations demonstrate the great physical robustness of the MFMR design.
3.3 Optimization of peak shapes
At high temperatures, gas viscosity increases, leading to flow restriction, and decreased flow rate in capillaries. Therefore, the relationship of MFMR hot zone channel diameter at 950°C furnace temperature, and He carrier gas head pressure (flow) was explored empirically by fabricating a series of MFMR. MFMR enclosures (pattern L275R0_65, as shown in Fig. 2b, with no metal reactant) of varying hot zone channel diameters were constructed.
An Agilent 6890 GC-FID system with electronic pressure control (EPC) was fitted with 1.7 m total length of 100 μm i.d. capillary to couple the MFMR with the GC inlet and FID. The dependence of CH4 FWHM peak widths on a wide range of head pressures was then explored. Flow rates of <1 ml/min to 6 ml/min were investigated depending on MFMR channel dimensions. Figure 4a shows that as EPC head pressure increases, peak widths decrease. FWHM peak widths of 219 ± 7 ms for 1.7 nmol CH4 are achievable for MFMR’s with pattern L275R0_65 and channel diameters of 155 and 185 μm, using head pressures greater than or equal to 55 psi. This is equivalent to the FWHM peak widths resulting from no MFMR in-line, 224 ± 8 ms, as shown in Table 1. For MFMR of pattern L275R0_65 at 950°C with hot zone channel diameters of 56 (not shown), 77, and 85 μm, peak widths of <250 ms were not attainable, even at the highest EPC head pressure (70 psi). Using these measurements, Fig. 4b demonstrates that fast GC-FID FWHM peak widths of less than or equal to 225 ms can be achieved with a MFMR of pattern L275R0_65 at 950°C composed of channel diameters approximately between 130 and 185 μm with head pressures greater than or equal to 55 psi (> 3 ml/min). Figure 5a depicts that the CH4 peak shape is the same through 1.7 m of 100 μm i.d. capillary at 25°C as through the same capillary length including the MFMR with 185 μm diameter channels at 950°C using FID detection. As a result, MFMR channel dimensions were successfully optimized so as to have a negligible effect on gas plug peak shapes at high temperatures.

CH4 gas was used to evaluate analyte peak broadening through MFMRs (pattern L275R0_65 with no reactant) with various channel diameters at various furnace temperatures and GC head pressures using He carrier gas in a fast GC-FID system. All MFMR hot zone channel lengths were 275 mm and channel diameters studied included 55 (not shown), 77, 85, 155, and 185 μm. In all cases, 2 μl of CH4 was injected at a 50:1 split, resulting in 1.7 nmol analyzed. An HP6890 GC-FID at various head pressures was used in (a) and (b), ranging from <1 to 6 ml/min, depending on head pressure and MFMR channel dimensions. The optimized channels of 155 and 185 μm, at head pressures >45 psi had flow rates of ~3–6 ml/min

Peak shapes of a 2 μl injections at 50:1 split, 1.7 nmol of CH4 through 1.7 m × 100 μm i.d. fused silica capillary without MFMR and with MFMR (L275R0_65Dc185 with no reactant) at 950°C using an HP6890 GC-FID with head pressure of 55 psi, and FID detection. Peak shapes of b 5 μl injections, at 100:1 split, 2 nmol of CO2 and CH4 through 2.5 m × 100 μm i.d. capillary, plus 0.5 m × 75 μm i.d. IRMS inlet capillary, without MFMR and with MFMR (L230R3Dc195Dw75 with oxidized metal reactant), with effective channel diameter <185 μm, using an HP6890 GC with head pressure of 50 psi, and IRMS detection of m/z 44 (MAT 252 IRMS). Data points are 40 ms apart (acquisition rate = 25 Hz). These are the peak shapes for the data presented in Fig. 6 and Table 1, and are achieved at a flow rate of ~3 ml/min
3.4 Combustion
A 55%Cu:45%Ni wire alloy (Constantan, Alfa Aesar, Ward Hill MA) compatible with 950°C combustion for 13C/12C analysis by IRMS was investigated. The pre-oxidized wire diameter of 75 μm was chosen so that the effective channel diameter, after insertion into the hot zone of MFMR’s with empty hot zone channel diameters of ~200 μm, was ~185 μm. After 100% oxidation, the wire volume should expand by almost a factor of two (~1.7), bringing the effective diameter to ~173 μm, targeting the middle of the optimized range (155–185 μm) for narrow peak shapes. However, the extent of oxidation and amount of expansion was not quantified in this work. MFMR’s with channel bend radii equal to or greater than 3 mm allowed reactant wire insertion; patterns L230R3 (Fig. 2e) and L150R4_1 (Fig. 2f) used in this study. The ports were then sealed to GC fused silica capillary tubing and the MFMR was interfaced to a fast-GCC-IRMS system. In order to prepare the MFMR for use as a combustion reactor, the hot zone region was held at 600°C using a high temperature furnace while oxygen was flowed through the MFMR for at least 24 h generating CuO and NiO. The oxidized 75 μm wire facilitated normal flows with narrow peak widths.
The peak widths and 13C/12C of CO2 and CH4 gas plugs were evaluated with and without an MFMR in line. An MFMR with pattern L230R3 (Fig. 2e) was used composed of a hot zone channel diameter of 195 μm containing a 75 μm diameter oxidized Constantan wire, resulting in an effective diameter of <185 μm (L230R3Dc195Dw75). The analysis of 2 nmol of CO2 without an MFMR was conducted to establish the baseline capability of the fast GCC-IRMS system. The analysis of 2 nmol of CH4 was conducted with the MFMR described above, held at 950°C. Results, in Table 1 and Fig. 5b, show that peak shapes and widths, which are ~250 ms FWHM, are not affected by the MFMR. However, this shows a little more broadening for the GCC-IRMS system compared to the GC-FID system (Fig. 5a), but is not unexpected due to the IRMS ion source inlet and the extra GC fused silica capillary length required for the GCC-IRMS configuration. Also, reproducibility of 13C/12C measurements of CH4 when using the MFMR was measured to be SD(δ13C) = ±0.28‰, which is the same as for plugs of CO2, which was SD(δ13C) = ±0.27‰. Figure 6 shows a plot of 10 consecutive injections of CO2 and CH4 and demonstrates no observed trend for 13C/12C values. This replicate analysis of CO2 and CH4 yielded similar m/z 44 peak areas, (1.1 ± 0.1 V s), suggesting completeness of CH4 oxidation since equivalent volumes of each gas contain equivalent moles of carbon.

The gases CO2 and CH4 were used to evaluate extent of combustion and reproducibility of 13C/12C isotope ratios produced by the MFMR. The 13C/12C (δ13C) measured for 10 consecutive injections of CO2 and CH4 are shown here. Injection volumes were 5 μl through the instrumental set up described in Fig. 5b. At a 100:1 split, this represents the analysis of approximately 2 nmol of each gas. 13C/12C’s were calculated relative to the 13C/12C of the first injection for each gas and is the same data summarized in Table 1
Our present system was evaluated with samples of approximately 2 nmol C, near the usual limit for precise and accurate IRMS analysis. In our previous study, samples of approximately 10 pmol C were considered, for which the counting statistics limit is SD(δ13C) ~ 0.3‰. We found that the precision scales inversely to the peak width. For instance, 250 ms FWHM peak widths from fast GCC-IRMS achieve 10 times better precision (0.4‰) than a 10-fold wider peak expected in normal GCC-IRMS (Sacks et al. 2007). Precision is likely to be limited by the ability to reproducibly define baseline, and shows that lower analyte concentrations can be employed while maintaining high precision because of the sharper peaks obtained with fast GC. Experiments addressing peak shape and combustion efficiency using the MFMR with different reactant types and quantities and channel dimensions for various compounds of interest, such as polar steroids, are required to characterize this property with MFMR. In these initial studies that focused on microfabrication, TorrSeal port seals were used and function only up to 120°C. Future experiments will be facilitated by the development of the MFMRs with, for instance, polyimide port seals that are resistant to GC oven temperatures up to 350°C, thus enabling compound separation analyses of lower vapor pressure and polar compounds, such as those with formula weights of >100 g/mol. Importantly, the MFMR can support larger channel dimensions for use as a more robust reactor as part of normal GCC-IRMS systems, improving their reliability as well.
4 Conclusions
Microfabricated micro-reactors (MFMR) were designed and developed using established and novel microfabrication techniques (Tobias and Brenna 2009). We adopted some process steps, to create circular channels, that resemble those of recent microfabrication of channels in various substrates (Bu et al. 2004; Grosse et al. 2001; Iliescu et al. 2008; Iliescu et al. 2005; Iliescu et al. 2007; Yu and Wing 1996). However, our processes were performed in high purity fused silica (SiO2), which is required for high temperature applications but also is a difficult material to micro-machine at the required dimensions of 100’s of micrometer and to maintain flatness and smoothness over many centimeter. Pure SiO2 has a much higher softening point and etches many times slower than impure SiO2 requiring well chosen protective layer masks for long etch times. Wet etching of small dimension structures (<20 μm), is routinely done and simpler compared to deep (>50 μm) wet etching due to the need for balance between etch time and protective layer resistance. Considering this, one particularly novel aspect to our fabrication process was the use of n-doped amorphous silicon (Si) which resisted HF etching well but could be removed quickly using KOH. It yielded a smooth substrate surface and routine success in creating permanently sealed devices after thermal bonding of wafers. Another original aspect of our process is a “two step isotropic wet etch” process to create tapered connection ports for input and output capillaries, which are similar to the commercially available Press-Tight® connections. Integrity of the reactor at elevated temperatures was assessed through leak checking. Fast GC plugs of CO2 gas and CH4 were used to evaluate peak broadening, extent of combustion, and 13C/12C reproducibility using the MFMR as part of a GCC-IRMS interface. In addition, hot zone channel bend radii equal to and greater than 3 mm allowed insertion of metal reactant wire. When interfaced to GCC-IRMS, the MFMR enables state of the art 13C/12C reproducibility, the hallmark of high precision IRMS.
References
Asche S, Michaud AL, Brenna JT (2003) Sourcing organic compounds based on natural isotopic variations measured by high precision isotope ratio mass spectrometry. Curr Org Chem 7:1527–1543
Brand WA (1995) US Patent No. 5,432,344
Brenna JT, Corso TN, Tobias HJ, Caimi RJ (1997) High-precision continuous-flow isotope ratio mass spectrometry. Mass Spectrom Rev 16:227–258
Bu MQ, Melvin T, Ensell GJ, Wilkinson JS, Evans AGR (2004) A new masking technology for deep glass etching and its microfluidic application. Sens Actuator A Phys 115:476–482
Burgoyne TW, Hayes JM (1998) Quantitative production of H2 by pyrolysis of gas chromatographic effluents. Anal Chem 70:5136–5141
Corso TN, Brenna JT (1997) High-precision position-specific isotope analysis. Proc Natl Acad Sci USA 94:1049–1053
Ellis L, Fincannon AL (1997) US Patent No. 5,783,741
Goodman KJ (1995) Hardware modifications to a VG isochrom yielding improved signal, resolution, and maintenance. 210th ACS National Meeting. American Chemical Society, Chicago, IL
Goodman KJ (1998) Hardware modifications to an isotope ratio mass spectrometer continuous-flow interface yielding improved signal, resolution, and maintenance. Anal Chem 70:833–837
Grosse A, Grewe M, Fouckhardt H (2001) Deep wet etching of fused silica glass for hollow capillary optical leaky waveguides in microfluidic devices. J Micromech Microeng 11:257–262
Hayes JM (1991) US Patent No. 5,012,052
Iliescu C, Jing J, Tay FEH, Miao JM, Sun TT (2005) Characterization of masking layers for deep wet etching of glass in an improved HF/HCl solution. Surf Coat Technol 198:314–318
Iliescu C, Tay FEH, Miao JM (2007) Strategies in deep wet etching of Pyrex glass. Sens Actuator A Phys 133:395–400
Iliescu C, Chen B, Miao J (2008) On the wet etching of Pyrex glass. Sens Actuator A Phys 143:154–161
Matthews DE, Hayes JM (1978) Isotope-ratio-monitoring gas chromatography-mass spectrometry. Anal Chem 50:1465–1473
Sacks GL, Zhang Y, Brenna JT (2007) Fast gas chromatography combustion isotope ratio mass spectrometry. Anal Chem 79:6348–6358
Sessions AL (2006) Isotope-ratio detection for gas chromatography. J Sep Sci 29:1946–1961
Tobias HJ, Brenna JT (1997) On-line pyrolysis as a limitless reduction source for high-precision isotopic analysis of organic-derived hydrogen. Anal Chem 69:3148–3152
Tobias H, Brenna JT (2009) US Patent No. (Application) 61/181,944
Tobias HJ, Sacks GL, Zhang Y, Brenna JT (2008) Comprehensive two-dimensional gas chromatography combustion isotope ratio mass spectrometry. Anal Chem 80:8613–8621
Yu CM, Wing CH (1996) US Patent No. 5,575,929
Acknowledgments
This work was supported by the United States Anti-Doping Agency (USADA) and the Partnership for Clean Competition (PCC)<http://www.cleancompetition.org/>. We thank Mike Skvarla, Rob Illic, and all other staff at the Cornell Nanoscale Science & Technology Facility (CNF) for instruction and assistance in microfabrication. CNF is supported by the National Science Foundation through the National Nanotechnology Infrastructure Network (Grant ECS-0335765). We would also like to thank Gavin Sacks and Larry Bowers for many helpful discussions.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Tobias, H.J., Brenna, J.T. Microfabrication of high temperature micro-reactors for continuous flow isotope ratio mass spectrometry. Microfluid Nanofluid 9, 461–470 (2010). https://doi.org/10.1007/s10404-010-0563-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10404-010-0563-x