
Summary
The heterogeneity of “rare bone disorders” can be explained by the number of molecules and regulatory pathways which are responsible for bone health and normal stature. In this article, the most important basic principles behind bone homeostasis from development to structure and regulation of the growing skeleton are summarized. The aim is to provide the reader with some theoretical background to understand the nature of the different main groups of disorders affecting bone stability, longitudinal growth and disturbances of calcium and phosphate homeostasis.
Zusammenfassung
Hinter der Gruppe seltener Knochenerkrankungen steht ursächlich eine Vielzahl unterschiedlicher Regulationsmechanismen, welche auch die Heterogenität der verschiedenen Erkrankungsbilder erklärt. In diesem Artikel soll ein Überblick über grundsätzliche Aspekte der Entwicklung, der Struktur und der Regulation des wachsenden Skelettsystems gegeben werden. Auf diese Weise sollen dem interessierten Leser die ganz unterschiedlichen seltenen Ursachen hinter erhöhter Frakturneigung, aber auch seltenen Wachstumsstörungen und Störungen der Mineralisation verständlich präsentiert werden.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The skeletal system of children is unique: Bone provides mechanical stability and at the same time allows longitudinal growth. These roles are established by rather separate “organs”, the bony tissue on one hand and the cartilaginous growth plate on the other hand. The growing bone of children and adolescents shows distinct characteristics when compared to adult bone and, as a matter of fact, growth is a process restricted to childhood and adolescence.
In this article we give an introduction on the structure and regulation of the growing skeleton with special emphasis on fundamental principles behind rare disorders of bone, growth and mineralization as a basis for their classification, diagnosis and treatments.
Bone and growth plate—basics
Development
In fetal life, limb buds appear during the 4th week of gestation, followed by rapid morphogenetic and segmentation processes. Already at the 8th week of gestation, large parts of the axial (thoracic cage, spine) and appendicular (extremities) skeleton are also mineralized at the sites of the primary ossification centers. At birth, there is a typical pattern of mineralized regions at the ends of long bones (secondary ossification centers). The growth plates are situated in between the primary and secondary centers of ossification.
The term ossification is central in the context of these important first steps in bone development. Ossification, meaning “the making of bones”, can either be established via a cartilaginous model (enchondral ossification) or directly from desmal origin (intramembranous ossification).
The term mineralization refers to all processes, where hydroxyapatite, the major mineral component of bone, is embedded in the respective matrix. Mineralization happens both in cartilaginous and bony matrices during physiological processes.
Structure
Bone is a unique tissue—besides its role of a connective and supportive tissue and hosting the hematopoietic system, bone serves as an important mineral reservoir and endocrine organ. During recent years enormous progress has been made in the field of bone research, in particular in the field of regulation of bone homeostasis under physiological and pathological conditions.
Bone and growth plate consist of a cellular and matrix component
Osteoblasts, the bone-forming cells arise from multipotent progenitor cells of mesenchymal origin. They pass a series of steps in differentiation and play the major role in bone formation by secretion of extracellular matrix proteins. Their ultimate fate is either to become a lining cell, differentiate into an osteocyte or undergo apoptosis [1].
Osteoblasts and osteoclasts communicate with each other to regulate bone homeostasis [2]. The RANK–RANKL–OPG system is the major regulatory system of osteoclast differentiation induction, activation, and survival [3].
Osteocytes previously often regarded as “dead cells embedded in a mineralized matrix” have only recently been recognized to be highly specialized regulatory cells [1]. Osteocytes have secretory roles in bone (e.g., fibroblast growth factor 23, FGF23) and are tightly connected in a mechano-sensing network responsive to intercellular and endocrine signaling.
Growth plate chondrocytes (Fig. 1a) are the masters of longitudinal growth [4]. Reserve cells (RC) enter a differentiation process along the long axis of bone. The number of proliferating chondrocytes (PC) as well as their differentiation into hypertrophic chondrocytes (HC) constitute the cellular contribution to growth. At the same time, cells secrete the surrounding matrix, which is typically avascular and unmineralized.

a Structure of the growth plate. b Local and systemic regulatory factors. RC reserve cells, PC proliferating cells, pHC prehypertrophic cells, HC hypertrophic cells, Ocl osteoclasts, Obl osteoblasts. Adapted from [10]
The extracellular matrix
The organic component of the extracellular matrix (ECM) of bone contains collagen (90%) and noncollagenous proteins (10%) [5]; Type I collagen is the most abundant protein of bone, its characteristic structure is a triple helix assembling in highly organized collagen fibrils. The abundant collagen of cartilage ECM is collagen type II (Fig. 1a). During enchondral ossification, the unmineralized matrix in the metaphysis around HCs becomes mineralized, is resorbed by osteoclasts and replaced by bone (reviewed in [6]).
Regulation
Local regulatory molecules and pathways
The local regulation of bone and cartilaginous tissues is performed by a multitude of complex pathways in constant interaction with external factors such as systemic and mechanical influences (see [7]).
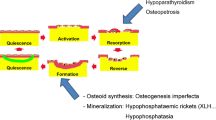
Bone tissue is in a continuous equilibrium of matrix formation and resorption. The coupling of the antagonistic processes is regulated by a complex system of local factors and molecular signals. A major role in intercellular communication is played by Receptor activator of nuclear factor-kappa B (RANK) and its ligand (RANKL). RANK, a member of the tumor necrosis factor family, is expressed by osteoclasts. Expression of RANKL by osteoblasts induces terminal osteoclast differentiation and keeps a balance of activity between these two antagonistic cell types. Modificators such as osteoblast-derived osteoprotegerin (OPG) interact in this regulatory system and allow adoption of the RANKL-driven coupling of osteoclasts and osteoblasts. In contrast to osteoclasts, osteoblast differentiation is mainly accomplished via three pathways: Bone morphogenic proteins (BMPs), Wnt signaling and Notch signaling. Many skeletal conditions such as fibrodysplasia ossificans progressiva or Hajdu–Cheney syndrome are caused by direct impairment of these regulatory systems. While novel insight into the regulation of this pathway, e.g., by microRNA expression, has been gained, the complex interaction of involved pathways is still under intensive investigation [7].
With respect to the growth plate (GP), studies in model systems, in vitro cultures and skeletal diseases have revealed an orchestration of transcription factors, soluble proteins, receptors and extracellular matrix components that regulate cellular processes and matrix metabolism (see [8]).
Several of the main players in the context of the regulation of enchondral ossification are depicted in Fig. 1. There is a unique feedback loop between Indian hedgehog (IHH) and parathyroid hormone-related protein (PTHrP) controlling terminal differentiation of HC and thus the height of the growth plate [9]. Fibroblast growth factors (FGFs) take another essential part of growth plate regulation, which is translated into genetic conditions associated with an overactivation of differentiation via the FGF-receptor 3 such as in achondroplasia.
The molecular mechanisms behind chondrocyte hypertrophy are complex [10]. SOX9 and RUNX2, which regulate transcription of collagen type X, matrix metalloproteinase 13 (MMP-13), vascular endothelial growth factor (VEGF) and IHH genes are involved (reviewed in [8]). MMP families are regarded as the most important degraders of ECM proteins and are known to play a part in the regulation of cell migration. VEGF, also produced by osteoclasts, appears to be the factor responsible for ingrowth of the vascular progenitor cells in the growth plate.
Local regulation of mineralization
The formation of hydroxyapatite crystals is tightly regulated to ensure sufficient matrix mineralization in hard tissues and to avoid ectopic mineralization. Pyrophosphates (PPis) represent a key regulator element being both potent inhibitors of mineralization and an essential source of phosphate for matrix formation. Secreted matrix vesicles (MV) containing PPi-degrading enzymes such as tissue-nonspecific alkaline phosphatase (TNSALP) play an essential role in the coordination of mineral nucleation [11, 12]. Short integrin-binding ligand-interacting glycoproteins (SIBLINGs) further regulate local phosphate homeostasis and link mineralization to systemic phosphate regulation, vascularization, energy metabolism, bone resorption and mechanotransduction [13].
Systemic regulation of bone and growth plate
Regulation of bone homeostasis via endocrine pathways is established on multiple levels [14, 15].
Parathyroid hormone (PTH) has a dual function in bone homeostasis: It supports bone formation by increasing osteoblast production and differentiation and inhibiting apoptosis, but also by locally stimulating the synthesis of IGF‑I. At higher serum concentrations, PTH stimulates osteoclast formation by increasing RANKL and reducing OPG synthesis. Both the active form of vitamin D (1,25-(OH)2 D), as well as its precursor (25-OH D) control bone remodeling by promoting both its formation and resorption. 25-OH D can be activated locally in osteoclasts and osteocytes via CYP27B1. Differentiation of osteocytes is directly regulated by 1,25-(OH)2 D via vitamin D receptors (VDR). Calcitonin seems to directly promote osteoclasts apoptosis. It also locally stimulates the synthesis of insulin-like growth factor 1 (IGF-1), thus supporting the proliferation and differentiation of osteoblasts. Similar to the growth plate, growth hormone also interferes with bone remodeling by directly stimulating osteoblast production and in addition, sustaining bone formation through local stimulation of IGF‑1 and IGF binding protein 3 (IGFBP-3) synthesis. Thyroid hormone was shown to directly act on both osteoblasts and osteoclasts but also indirectly through cytokines. It has a dual effect in osteoblasts, as it can stimulate the differentiation, but also inhibit their proliferation. T3 also seems to promote osteoclastic activity. Estrogens seem to play a role in controlling apoptosis in bone cells (inhibition in osteoblasts, stimulation in osteoclasts). The production and the activity of osteoclasts is inhibited by estrogens by downregulating the production of RANKL and by stimulating the synthesis of OPG. Androgens were shown to indirectly downregulate osteoclastic activity by its actions on the RANKL/RANK/OPG system, thus playing a role in preserving bone mass. Glucocorticoids (GCs) directly and indirectly influence bone homeostasis by decreasing intestinal calcium absorption and increasing its urinary excretion. Due to the inhibition of gonadotropin secretion, it negatively influences bone formation. GC receptors were found in osteoblasts, so it has been theorized that GCs directly inhibit the formation and differentiation of osteoblasts but might also stimulate their apoptosis. Furthermore, GCs seems to sustain bone resorption by stimulating RANKL synthesis and inhibiting OPG secretion, thus promoting osteoclasts formation.
Multiple systemic hormones play a role in the regulation of the growth plate homeostasis, such as thyroid hormones, estrogens, vitamin D, but especially serum IGF‑I and GH [16].
Estrogen receptors were found in the growth plate, the exact mode of action is still under debate although an effect on chondrocyte senescence has been proposed [17]. Many of the effects of systemic androgens are locally mediated via aromatization into estrogens, such as bone age maturation and growth plate fusion, but presence of androgen receptors in the growth plate also suggest direct actions. Thyroid hormones seem to activate the Wnt/beta-catenin signaling pathway and, as a result, stimulate the differentiation of chondrocytes. T3 is known to also promote local production of IGF‑1, which in turn is involved in chondrocyte hypertrophy.
The somatomedin hypothesis had to be adjusted over time, in more recent years both direct and indirect effects of GH and IGF‑I on the growth plate as well as synergistic and antagonistic effects of GH and IGF‑I on metabolism and regulation of bone metabolism have been recognized [18].
Main groups of rare bone and growth disorders
Classification in subcategories of rare bone disorders can be made either according to the affected tissue component or dominating clinical symptomatology. Nosologies have increasingly moved from radiological to molecular [19] or regulatory based classifications [20].
When it comes to clinical practice, the following groups of rare bone and growth disorders are predominant:
In regard to matrix components and mechanical properties of bones, osteoporotic and rachitic conditions can be distinguished: Osteoporotic conditions are characterized by an increased bone fragility and high fracture rates, while rachitic disorders feature matrix impairment leading to decreased stiffness and development of long bone deformities. Conditions associated with increased formation of bone matrix are defined as osteosclerotic disorders and can lead to impaired biomechanical matrix properties by irregular architecture rather than by lack of synthesis.
Bone fragility disorders
Osteoporosis in pediatric patients represents a severe condition defined by impaired bone structure and increased bone fragility. Primary osteoporosis is caused by genetic deviations in bone matrix or regulator genes and has to be distinguished from secondary or acquired osteoporosis, which represent the most common cause for bone fragility in pediatrics [21]. Osteogenesis imperfecta (OI, OMIM #166200) describes a group of heterogeneous, genetic conditions and constitutes most cases of primary bone fragility. The phenotypic spectrum varies from a moderately increased fracture rate in late adulthood to perinatal death due to intrauterine fractures and deformities [22]. Mutations in collagen type I (COL1A1 or COL1A2) represent the most frequent alteration in patients with OI, constituting approximately 80% of cases. Less common OI types involve regulatory genes for collagen structure, synthesis, folding, secretion and matrix organizations (Table 1).
Sclerosing bone disorders
Sclerosing bone disorders are rare genetic conditions defined by abnormally increased deposition of mineralized matrix [23]. Most disorders are caused by pathologic mechanisms involving endochondral or intramembranous ossification, such as juveniles Paget’s disease, van Buchem disease and pycnodysostosis. Despite increased bone density, some disorders are associated with increased fracture rates due to irregular bone matrix. Cranial involvement is often associated with severe neurological symptoms due to nerve compression.
Rickets and osteomalacia
Rickets are characterized by dysregulation of growth plate cartilage caused by mineral deficiency. After cessation of growth the term osteomalacia is used. While most cases of rickets are caused by vitamin D or calcium deficiency, hereditary hypophosphatemic rickets comprises a group of orphan diseases characterized by renal phosphate wasting due to dysregulation of the main endocrine regulator of phosphate handling, FGF23. X‑linked hypophosphatemic rickets (XLH) represents the most common type of hypophosphatemic rickets with a prevalence of approximately 1 in 20,000 and is caused by loss-of-function of PHEX. In XLH, excess FGF-23 reduces renal phosphate reabsorption and vitamin D activation, resulting in chronic hypophosphatemia. Impaired growth and severe rickets are major characteristics of XLH, representing the main causes of morbidity and surgical interventions in these conditions. Onset of symptoms is commonly observed at the beginning of gait due to incremental deformities of weight bearing bones [24].
Rare disorders of PTH and vitamin D
Rare hypercalcemic disorders
Hypercalcemia can be seen in various rare genetic disorders, with different PTH levels [25, 26]: Primary hyperparathyroidism (PHPT), due to glandular hyperplasia or adenomas, is rare during childhood. Hereditary familial forms of hyperparathyroidism (HPT) are multiple endocrine neoplasia (MEN) and hyperparathyroidism-jaw tumor syndrome (HPZ-JT). The non-syndromic forms of HPT and their respective mutated genes are familial isolated hyperparathyroidism (FIHP: MEN1, PTH and GCM2), neonatal severe primary hyperparathyroidism (NSHPT: homozygous or compound heterozygous inactivating mutations in the CaSR) and familial hypocalciuric hypercalcemia (FHH: CaSR). The following rare disorders are associated with low PTH levels: Jansen metaphyseal chondrodysplasia (activating mutations in PTHR1), Williams Beuren syndrome (multiple gene deletions of chromosome 7q11.23) Inactivating mutations of CYP24A1 were found in patients diagnosed with idiopathic infantile hypercalcemia and explain the pathomechanism: vitamin D is not catabolized and it accumulates in serum, resulting in an increased intestinal calcium absorption [25,26,27].
Rare hypocalcemic disorders
Hypocalcemia is either caused by reduced PTH synthesis or impaired action, as well by deranged activity of CaSR and by disorders of vitamin D metabolism; in the pediatric population, hypocalcemia often is caused by a genetic disorder [26]. Isolated hypocalcemia can be found in agenesis (GCM2 mutations) or in autoimmune destruction of the parathyroid glands, or in disorders leading to dysfunctional synthesis/bioactivity of PTH by PTH mutations. Some of the complex genetic pathologies and the respective genes causing hypocalcemia are DiGeorge syndrome (TBX1 or NEBL), autoimmune polyglandular syndrome type 1 (AIRE), CHARGE syndrome (CHD7, SEMA3E), Kenney–Caffey syndrome (TBCE, FAM111A), hypoparathyroidism, sensorineural deafness and renal syndrome (GATA3). Familial cases, with an autosomal dominant, recessive or X‑linked inheritance were also described in the literature. An impaired secretion of PTH is seen in activating mutations of the CaSR, as in autosomal dominant hypocalcemia type 1 (ADH1). Some cases present with severe hypocalcemia, while others remain asymptomatic. High serum PTH levels, due to PTH resistance, is characteristic for pseudohypoparathyroidism (PHP; new nomenclature: inactivating PTH/PTH-related protein signaling disorders iPPSD). In 4 out of the 6 iPPSDs, PTH resistance has been described so far: type 2 (includes former PHP1a and 1c) due to Gs α inactivating mutations, type 3 (PHP1b) caused by methylation defects of GNAS DMRs (differentially methylated regions), type 4 (acrodysostosis type 1) through mutations of PRKAR1A and type 5 (acrodysostosis type 2) caused by mutated PDE4D [28]. Hypocalcemia is also found in vitamin D-dependent rickets type 2, an autosomal recessive disorder caused by mutated vitamin D receptor (VDR), as well as in vitamin D-dependent rickets type 1, due to impaired 1‑α hydroxylation (CYP27B1 mutations).
Rare growth disorders
Both molecular and endocrine dysfunctions can lead to either attenuated or accelerated linear growth resulting in short or tall stature. Disproportionate short stature is found in most subgroups of skeletal dysplasias, but molecular defects within the growth plate or connective tissue can also lead to tall stature, e.g., in Marfan syndrome. A comprehensive survey on skeletal dysplasias is given in Walleczek et al., this issue. Severe growth disorders affecting the GH/IGF‑I axis have been reviewed recently [29].
Diagnosis of rare bone and growth disorders
Bone fragility disorders
Pediatric osteoporosis is defined by a clinical significant fracture history alone or in combination with impaired bone mineral density [30]. In order to identify primary bone disorders at an early stage, a more differentiated approach including typical clinical features of osteogenesis imperfecta or hypophosphatasia has recently been proposed [31]. On suspicion, specialized clinical and lab assessment, lateral spine imaging for vertebral fractures and, if indicated, genetic analyses for OI are recommended to reduce diagnostic delays.
Rickets
Comprehensive assessment for rickets aims to distinguish vitamin D deficiency from rare genetic types such as XLH or renal causes, e.g., Fanconi syndrome. In the clinical presence of rickets, relatively normal 25(OH)D values despite increased low age specific serum phosphate values are typical findings in XLH. Further diagnostics include urinary analysis (TmP/GFR, Fanconi assessment), ALP, PTH and—if available—intact serum FGF23. All patients with suspected genetic types of hypophosphatemic rickets should undergo genetic diagnostics [24].
Growth disorders
The measurement of body proportions helps to direct diagnostics to first-line endocrine (proportional short stature) or non-endocrine reasons for short stature. Disproportionate growth disorders, together with symptoms affecting the skeletal system or connective tissue point to a genetic disorder and prompt genetic analysis and radiological investigations (see Walleczek et al. this issue and Mehany and Patsch, this issue). Comprehensive reviews on the diagnostic work-up are available [32].
Role of bone biopsy in rare bone disorders
Many clinically important conclusions have been drawn from bone biopsies during clinical studies in OI and other rare bone diseases. Sophisticated techniques help to evaluate bone material quality but also aspects of mineralization, as reviewed by Mähr et al. in this issue.
Therapies
Detailed treatment recommendations are beyond the scope of this article (we refer to Kocijan in this issue and recently published consensus guidelines). A few important principles referring to rare pediatric bone disorders are summarized as follows:
Pharmacological therapies
Bone fragility
Pharmacologic interventions for pediatric osteoporosis is predominantly based on bisphosphonates. Data derived from patient cohorts with OI could show a clear improvement of bone mineral density as well as reduction of skeletal pain. While data on bone fracture rates is not entirely clear, bisphosphonates are standard of care for pediatric patients with OI and are increasingly applied for other indications such as secondary osteoporosis, giant cell aneurysm or chronic recurrent multifocal osteomyelitis [33].
Hypophosphatemic rickets
Conventional treatment of hypophosphatemic rickets consists of oral phosphate salts and active vitamin D derivatives. Due to its short half-life time, phosphate has to be given in 3–6 doses/day, leading to frequent compliance issues. Treatment has to be initiated at optimum in the neonatal period for an optimized clinical benefit. Global approval of an FGF23 blocking antibody opened a novel therapeutic perspective for patients with XLH. Subcutaneous application of burosumab reduces renal phosphate excretion and normalizes serum phosphate in a majority of XLH patients. Nevertheless, both treatment options are only improving the phenotype with persisting growth restriction and unclear effects on dental and cranial symptoms [24].
Hyper- and hypocalcemia
Acute hypo- and hypercalcemia are emergencies and should be treated according to published recommendations [25, 26, 34]. One should take in consideration the underlying disorder when substituting calcium and vitamin D; for CaSR mutations, this substitution should be reserved for the symptomatic phases.
The multidisciplinary team
Most complex skeletal disorders typically affect many organ systems, thus, stressing the need for coordinated multidisciplinary care. Osteology, orthopedics, radiology, neurology and neurosurgery, functional therapies and psychology are among the main disciplines involved in long-term care for this vulnerable patient group.
Specialized orthopedic treatments are an essential component of multidisciplinary care in pediatric bone disorders. Specialized assessment, well-coordinated planning of interventions and conservative treatments have to be adapted to the conditions’ characteristics and individual patient’s needs (reviewed in Mindler et al. this issue, Willeger et al. this issue).
Measures of success
Initiatives such as European Reference Networks facilitated the formation of expert centers able to provide sufficient structures to ensure high-quality, cost-effective multidisciplinary care. Strong cooperation with patient groups as well as the transnational exchange and clinical studies represent key elements of future individualized patient care and will help to improve life quality and disease awareness. In this context, patient registries, definitions of measures of success by evidence-based instruments with a focus on patient reported outcomes is essential. This includes parameters of inclusion and participation, mobility, reduction of pain, comprehensive quality-of-life measures adapted to the respective condition, and identification of treatment goals.
References
Robling AG, Bonewald LF. The Osteocyte: new insights. Annu Rev Physiol. 2020;82(1):485–506.
Kim J‑M, Lin C, Stavre Z, Greenblatt MB, Shim J‑H. Osteoblast-osteoclast communication and bone homeostasis. Cells. 2020;9(9):2073. https://doi.org/10.3390/cells9092073.
Udagawa N, Koide M, Nakamura M, Nakamichi Y, Yamashita T, Uehara S, et al. Osteoclast differentiation by RANKL and OPG signaling pathways. J Bone Miner Metab. 2020. https://doi.org/10.1007/s00774-020-01162-6.
Hunziker EB. Mechanism of longitudinal bone growth and its regulation by growth plate chondrocytes. Microsc Res Tech. 1994;28(6):505–19.
McKee M, Cole W. Bone matrix and mineralization. In: Glorieux F, Pettifor J, Juppner H, editors. Pediatric Bone. Biology and Diseases. Amsterdam: Elsevier; 2012. pp. 9–37.
Kronenberg HM. Developmental regulation of the growth plate. Nature. 2003;423(6937):332–6.
Zaidi M, Yuen T, Sun L, Rosen CJ. Regulation of skeletal homeostasis. Endocr Rev. 2018;39(5):701–18.
Hallett SA, Ono W, Ono N. Growth plate chondrocytes: skeletal development, growth and beyond. Int J Mol Sci. 2019;20(23):6009. https://doi.org/10.3390/ijms20236009.
Chung U, Lanske B, Lee K, Li E, Kronenberg H. The parathyroid hormone/parathyroid hormone-related peptide receptor coordinates endochondral bone development by directly controlling chondrocyte differentiation. Proc Natl Acad Sci U S A. 1998;95(22):13030–5.
Haeusler G, Raimann A, Egerbacher M. Growth plate research. In: Pietschmann P, editor. Principles of bone and joint research. Berlin Heidelberg: Springer; 2017. pp. 153–71. Learning Materials in Biosciences.
Golub EE. Role of matrix vesicles in biomineralization. Biochim Biophys Acta. 2009;1790(12):1592–8.
Orimo H. The mechanism of mineralization and the role of alkaline phosphatase in health and disease. J Nippon Med Sch. 2010;77(1):4–12.
Rowe PSN. Regulation of bone–renal mineral and energy metabolism: the PHEX, FGF23, DMP1, MEPE ASARM pathway. Crit Rev Eukaryot Gene Expr. 2012;22(1):61–86.
Florencio-Silva R, Sasso GRS, Sasso-Cerri E, Simões MJ, Cerri PS. Biology of Bone tissue: structure, function, and factors that influence bone cells. Biomed Res Int. 2015;2015:421746. https://doi.org/10.1155/2015/421746.
Siddiqui JA, Partridge NC. Physiological bone remodeling: systemic regulation and growth factor involvement. Physiology. 2016;31(3):233–45.
van der Eerden BCJ. Systemic and local regulation of the growth plate. Endocr Rev. 2003;24(6):782–801.
Lui JC, Nilsson O, Baron J. Growth plate senescence and catch-up growth. Endocr Dev. 2011;21:23–9.
Kaplan SA, Cohen P. REVIEW: the Somatomedin hypothesis 2007: 50 years later. J Clin Endocrinol Metab. 2007;92(12):4529–35.
Mortier GR, Cohn DH, Cormier-Daire V, Hall C, Krakow D, Mundlos S, et al. Nosology and classification of genetic skeletal disorders: 2019 revision. Am J Med Genet A. 2019;179(12):2393–419.
Etich J, Rehberg M, Eckes B, Sengle G, Semler O, Zaucke F. Signaling pathways affected by mutations causing osteogenesis imperfecta. Cell Signal. 2020;76:109789.
Saraff V, Högler W. Osteoporosis in children: diagnosis and management. Eur J Endocrinol. 2015;173(6):R185–97.
Van Dijk F, Sillence D. Osteogenesis imperfecta: clinical diagnosis, nomenclature and severity assessment. Am J Med Genet A. 2014;164(6):1470–81.
Boulet C, Madani H, Lenchik L, Vanhoenacker F, Amalnath DS, de Mey J, et al. Sclerosing bone dysplasias: genetic, clinical and radiology update of hereditary and non-hereditary disorders. Br J Radiol. 2016;89(1062):20150349. https://doi.org/10.1259/bjr.20150349.
Haffner D, Emma F, Eastwood DM, Duplan MB, Bacchetta J, Schnabel D, et al. Clinical practice recommendations for the diagnosis and management of X‑linked hypophosphataemia. Nat Rev Nephrol. 2019;15(7):435–55.
Stokes VJ, Nielsen MF, Hannan FM, Thakker RV. Hypercalcemic disorders in children. J Bone Miner Res. 2017;32(11):2157–70.
Allgrove J, Shaw NJ, editors. Calcium and Bone disorders in children and adolescents. Book: Karger; 2015.
Bringhurst FR, Demay MB, Kronenberg HM. Hormones and disorders of mineral metabolism. In: Williams textbook of endocrinology. Amsterdam: Elsevier; 2011. pp. 1277–94.
Thiele S, Mantovani G, Barlier A, Boldrin V, Bordogna P, De Sanctis L, et al. From pseudohypoparathyroidism to inactivating PTH/PTHrP signalling disorder (iPPSD), a novel classification proposed by the EuroPHP network. Eur J Endocrinol. 2016;175(6):P1–17.
Jee YH, Baron J, Nilsson O. New developments in the genetic diagnosis of short stature. Curr Opin Pediatr. 2018;30(4):541–7.
Pediatric Positions. ISCD.. https://iscd.org/learn/official-positions/pediatric-positions/. Accessed 14 Nov 2020.
Ward LM, Weber DR, Munns CF, Högler W, Zemel BS. A contemporary view of the definition and diagnosis of osteoporosis in children and adolescents. J Clin Endocrinol Metab. 2020;105(5):e2088–97.
Collett-Solberg PF, Ambler G, Backeljauw PF, Bidlingmaier M, Biller BMK, Boguszewski MCS, et al. Diagnosis, Genetics, and Therapy of Short Stature in Children: A Growth Hormone Research Society International Perspective. Horm Res Paediatr. 2019;92(1):1–14.
Simm PJ, Biggin A, Zacharin MR, Rodda CP, Tham E, Siafarikas A, et al. Consensus guidelines on the use of bisphosphonate therapy in children and adolescents. J Paediatr Child Health. 2018;54(3):223–33.
Davies JH. A practical approach to problems of Hypercalcaemia. Calcium Bone Disord Child Adolesc. 2009;16:93–114.
Author information
Authors and Affiliations
Contributions
GH initiated the review, AR, DAE and GH contributed equally in literature research, drafting and writing of the manuscript. After critical final review, all authors gave their consent to submit the paper.
Corresponding author
Ethics declarations
Conflict of interest
A. Raimann, D.-A. Ertl and G. Haeusler declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
A. Raimann and D.-A. Ertl are co-first authors.
Rights and permissions
About this article

Cite this article
Raimann, A., Ertl, DA. & Haeusler, G. Bone and growth: basic principles behind rare disorders. Wien Med Wochenschr 171, 86–93 (2021). https://doi.org/10.1007/s10354-020-00809-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10354-020-00809-3