
Abstract
Linearity of response and sensitivity (i.e., gradient of the calibration curve) were selected as performance parameters for the comparison of six GC–MS methods that result from all possible combinations of the two ionization (electron ionization, EI, and negative chemical ionization, NCI) and the three mass scanning techniques (full scan, selective ion monitoring and MS–MS). The study involved testing of spiked matrix (tomato) samples as well as standard solutions of 27 pesticides in acetonitrile at a range of concentrations (10–200 μg L−1). The selected pesticides are representative of some of the most common pesticide classes. The performance of each technique was quantified and a comparison made between the different methods. MS/MS was found to perform better as far as linear response was concerned, especially in plant matrix, in agreement with scientific literature. Sensitivity was enhanced with NCI–full scan and EI–MS/MS.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Gas and liquid chromatographs coupled with single or triple quadrupoles or ion trap mass spectrometric detectors are the most widely used analytical tools for routine analysis [1, 2]. In the past decade, liquid chromatography combined with triple quadrupole spectrometers (LC–MS/MS) has become the method of choice in the field of routine analysis for pesticide residues in food and environmental samples [3–10]. Electrospray ionization (ESI) is the most predominant ionization technique in LC–MS/MS. On the other hand, electron impact (EI) ionization remains the most popular technique for GC–MS and GC–MS/MS, compared to chemical ionization (CI).
GC–MS/MS did not receive initially the wide acceptance one would expect from the fact that historically GC–MS was the first mass spectrometric technique widely used in the field of pesticide residues analysis [11]. This could be attributed to the limitations in GC–MS/MS that arise from the absence of a universal soft ionization mode, which could be used for the efficient production of molecular ions of most pesticide classes. [11] EI ionization is more universal, but often the total ion current is spread on many fragments, resulting in a low intensity of precursor (parent) ions of MS/MS experiments, thus affecting not only the sensitivity but also the specificity of the method [1, 11]. Even so, a good suppression of matrix background is obtained by GC–MS/MS systems, in analogy to CI–MS and GC–TOF [11].
Negative chemical ionization (NCI) being a “softer” ionization technique generates high-intensity ions (in most cases molecular ions) of only some pesticide classes [12–14]. In combination with MS/MS, NCI gives better selectivity for compounds with highly electronegative elements such as halogen, oxygen and nitro group. This results in chromatograms with reduced matrix interference [1]. However, in single quadrupole GC–MS it was observed that the signal intensity of different pesticides (if identical amounts are injected) varies much more compared to EI ionization. Consequently, GC–MS with chemical ionization has been focused on special substance classes only, for example, organohalogen pesticides, pyrethroids and organophosphates. It has been rarely used in multi-residue methods, because it is not a universal ionization technique [11]. However, the initial problem that the mass spectra produced by chemical ionization usually contain a smaller number of fragments thus offering less information compared with EI has been overcome with the use of the QqQ mass analyzer. A number of methods have been developed in the past 15 years for the determination of pesticides or other pollutants such as polyaromatic hydrocarbons and polychlorinated biphenyls using gas chromatography coupled with EI–MS [15], EI–MS/MS [16], NCI–MS [17, 18], NCI–MS/MS [19].
Generic sample preparation leading to sample extracts that are analyzed by both LC–MS/MS and GC–MS/MS is nowadays applied in pesticide residue analysis (PRA) [1]. A combination of GC–MS/MS and LC–MS/MS is currently one of the most powerful approaches in PRA. They are complementary techniques that allow the determination of pesticides and metabolites within the whole range of physico-chemical properties, such as volatility, polarity and thermal stability. The combined use of both techniques allows the monitoring of hundreds of compounds that are GC or LC amenable [1].
The present study involves the comparison of performance characteristics (linearity of response, sensitivity, i.e., gradient of the calibration curve) [20] of six GC–MS methods that result from all possible combinations of two ionization (electron ionization and negative chemical ionization) and three mass scanning techniques (full scan, selective ion monitoring and MS–MS) applied in both plant matrix and solvent for PRA. Although there is extensive literature presenting applications of each of the above methods separately as well as their advantages and disadvantages, there is no publication presenting a comparison between the six methods based on the quantification of performance parameters. We aim to fill this gap with our current study.
Materials and Methods
The analytical standards of the pesticides were provided by Dr. Ehrenstorfer (Darmastaad, Germany) and were escorted with the appropriate certificate of analysis. Solvents were of HPLC grade (Fisher Scientific UK Limited, Loughborough, UK, Merck, Darmstadt, Germany).
The instrumentation used for the comparisons was a Varian 3800 gas chromatograph (Bruker Corporation, Fremont California) with a large volume programmable temperature vaporization (PTV) injector, coupled to a Varian 1200L triple quadrupole mass spectrometer (Bruker Corporation, Fremont California).
Selection of Pesticides
The 27 pesticides selected for this study were representative of the most common pesticide classes such as pyrethrinoids, organophosphorus, organochlorines, chlorophenyl, pyrimidine, diphenyl ether, dinitroaniline, triazoles and dicarboximide. To ensure the flexibility of the scope, the selection was also made according to their physicochemical properties to cover a wider range of different values of octanol–water constant (K ow), water solubility and vapor pressure. Due to the relatively small number of pesticide groups that are GC amenable, the variation of these values is rather limited. However, based on EU guidance documents [21] the selection of representative analytes can be used to extrapolate validation data to a whole chemical group.
Preparation of Standard Solutions
The stock solutions of the individual pesticide standards were prepared by accurately weighing 10–50 mg of each analyte in certified ‘A’ class volumetric flasks and dissolving in acetone. The stock standard solutions were stored at −20 °C. A single composite working standard solution was prepared by combining aliquots of each stock solution and diluting in acetonitrile to obtain a final concentration of 1 mg L−1. The working standard solution was also stored at −20 °C and before each use was left to reach room temperature. Two series of calibration standards were prepared within the range 10–200 μg L−1 by serial dilution in acetonitrile and tomato extract.
Preparation of Matrix Extract
The extraction procedure used for the preparation of the matrix extract was based on the “QuEChERS” method protocol for fruits and vegetables [22]. A 10 g portion of homogenized tomato was weighed in a 50 mL PTFE centrifuge tube, 10 mL of acetonitrile was added and the tube was vigorously shaken for 1 min. A mixture of 1 g of NaCl, 4 g of MgSO4, 1 g of trisodium citrate dehydrate and 1 g of disodium hydrogen citrate sesquihydrate was added and the tube was vigorously shaken for 1 min or more to prevent coagulation of MgSO4. The sample was then centrifuged at 4,000 rpm for 5 min. An aliquot of 6 mL of the acetonitrile phase was transferred into a 15 mL centrifuge tube containing 150 mg of PSA and 900 mg of MgSO4. The tube was shaken vigorously for 1 min and centrifuged for 5 min at 4,000 rpm. An aliquot of 5 mL of the cleaned-up extract was transferred into a screw cup storage vial, taking care to avoid sorbent particles from being carried over. The extract was used for the preparation of matrix-matched calibration standards by serial dilutions as described above.
Gas Chromatography
Aliquots of 5 μL of sample extract were injected into the gas chromatograph. The initial injector temperature 90 °C was held for 0.75 min and then increased at 200 °C min−1 to 280 °C, which was held for 5 min. The injector split ratio was first set at 30:1. After 0.75 min, splitless mode was set until minute 3. At 3 min, the split ratio was set at 60:1 and at 6 min the split ratio was 30:1. The capillary column used for the separation of the analytes was a Varian Factor Four VF-5 MS (30 m × 0.25 mm ID × 0.25 μm film thickness) with a deactivated cyano-phenyl-methyl guard column (5 m × 0.53 mm ID). The GC oven temperature program started from 70 °C (2 min), increased with 30 °C min−1 to 180 °C, then with 1.8 °C min−1 to 230 °C and finally with 30 °C min−1 to 280 °C (30 min). The temperatures of the manifold and the transfer line were 280 and 40 °C, respectively, and the electron multiplier was set at 1500 V.
Ionization
With electron ionization (EI), the ion source parameters were the following: ion source temperature 250 °C, filament current 50uA, electron energy 70 eV. With negative chemical ionization, the ion source temperature was 180 °C, CI gas pressure 6.2 Torr, filament current 50 uA and electron energy −70 eV. The source temperature in both cases of EI and NCI were determined after optimization study for the best signal of the analytes.
Mass Spectrometry
The parameters for the three mass spectrometric modes used were: (a) full scan (MS) with scanning range 50–1,000 amu and dwell time 1 min, (b) selective ion monitoring (SIM) using only the first quadrupole for mass separation, (c) multiple reaction monitoring (MRM) using Q1 and Q3 for mass separation and Q2 as a collision cell filled with Ar at 1.5 mbar. \( S_{u} = \frac{{S_{{{\text{Area}}/C}} }}{b} \) The MS and MS/MS optimization was conducted for both CI and EI mode separately, since the ions formed in each case are not identical in most cases. During the optimization procedure, multiple injections of each analyte at a concentration of 100 μg L−1 and different electron energy and collision energy values were preformed. MS and MS/MS spectra were acquired to obtain information about the maximum abundance of each ion or ion transition for each compound.
In both CI and EI modes, the optimum electron energy was 70 eV and the optimum collision energy between 5 and 70 eV, depending on the analyte. The retention times (RT), ions and transitions of each analyte are presented in Table 1.
Experimental Design
To estimate the effect of the three variable parameters (matrix, ionization and MS technique) on the key performance characteristics under study (linearity, sensitivity), a series of calibration standards in solvent and a series of calibration standards in matrix extract were injected in the chromatographic system under the same conditions, as summarized in Table 2.
For data processing of the chromatograms, the Varian MS Workstation software version 6.8 was used, and for statistical analysis of the data Microsoft Excel 2007. The statistical evaluation of the data was conducted using the peak area of each peak.
To verify the accuracy of the measurements, control chart monitoring during the whole experimental period was preformed. The control charts were constructed by injecting for 15 days a quality control standard solution (QCS) of the analytes diazinon, chlorpyrifos ethyl, α-endosulfan and lambda-cyhalothrin at 10 μg L−1 in acetonitrile. The retention time, ion ratio and peak area were recorded and for each parameter the control limits were set at 2 and 3 times the standard deviation(s). To ensure the system suitability before the beginning of the experiment and every ten injections, a QCS was injected. No exceedance above 3 s was observed and therefore the measurements are considered to be valid.
Linearity
Linearity of the methods was evaluated at five concentration levels: 10–25–50–100 and 200 μg L−1.
Pair Comparisons: Significance of Influence of Parameters on Sensitivity
To study if the two different ionization modes (NCI and EI) significantly influence the sensitivity (i.e., gradient of the calibration curve) of the detector to the analytes, the slopes of the calibration lines obtained by each ionization mode were compared using a Student’s t test. In the same way, the three different MS modes (full scan, SIM and MRM) were compared as well as the two different matrices (solvent and tomato).
The calculated value t cal used for the comparisons is defined in the following equation:
with b 1 and b 2 the slopes of the two calibration lines compared and S b1and S b2 the standard deviation of the slopes. If the calculated value t cal is smaller than the theoretical value (t theo) of 2,306 (df = 5 + 5 − 4 = 6, two-sided critical region, probability 95 %), then there is no significant difference between the two calibration lines (accepted null hypothesis).
Pair Comparisons: Estimation of Magnitude of Difference
To estimate the magnitude of the difference between two parameters, paired comparison between the slopes of the calibration lines of these parameters were performed. The null hypothesis is that the effect of this parameter is not significant. The magnitude of the difference (diff) expressed in % sensitivity enhancement or reduction [24] was estimated as:
where the parameter factor (PF) is calculated with the following equation:
and b 1 and b 2 are the slopes of the calibration lines of the analytes, respectively.
Results and Discussion
Linearity
As shown in Fig. 1, in most cases the analytes presented acceptable linearity with correlation coefficients (r) better than 0.95. In solvent, CI–SIM and EI–MS/MS seem to perform better for r > 0.98. However, in matrix it is only EI–MS/MS that retains good performance and CI–MS/MS seems to perform better in matrix than in solvent. The poorest linearity was observed in the cases of EI–full scan and EI–SIM when the analytes were in matrix. The results are in accordance with the findings of Huskova et al. [13] who reported better performance in PRA for CI–SIM compared to EI–SIM, with better linearity expressed as correlation coefficient r as well as better sensitivity and repeatability.

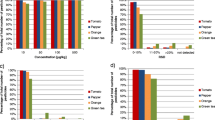
Linearity in solvent (a) and matrix (b): axis y represents the percentage of analytes that exhibited better correlation coefficient r than the figure in axis x. The total percentage is given for each figure for the differences between the techniques to be more apparent
With EI–MS–full scan, the percentage of the analytes with r values below 0.9 was 29 % in solvent and 36 % in matrix. With EI–SIM the corresponding values were 14 and 43 %. It is obvious from Fig. 1 that CI–MS/MS and EI–MS/MS perform better in matrix with a linearity correlation coefficient r better than 0.95 for 75 and 79 % of the tested pesticides, respectively. However, it must be underlined that EI–MS/MS is the only method that performs almost equally in both matrix and solvent as far as correlation coefficients better than 0.99, 0.98 and 0.95 are concerned, implying robustness to matrix effect.
Statistical Evaluation of the Results
Significant differences in sensitivity were observed between the ionization modes, MS modes and the two matrices for most of the analytes. The MS technique (95.2 %), the ionization (91.1 %) and the matrix (57.2 %) were found to affect significantly the sensitivity of the methods. The variation due to the presence of the matrix is observed in approximately half (57.2 %) of the comparisons preformed, in contrast to the other parameters for which the variation is significantly higher in almost the total of the comparisons conducted. In most cases for organochlorines (69.6 %), no significant matrix effect was observed. These percentages are lower for organophosphorus (42.1 %), pyrethrinoids (50 %) and for other chemical classes (49.2 %).
Comparison of MS Determination Techniques
To estimate the significance of the effect of the MS technique on sensitivity, the three different techniques (full scan, SIM and MS/MS) were compared in pairs and the differences were estimated as % sensitivity increase or decrease. In Fig. 2, a graphical presentation of the sensitivity variations from pair comparison of the MS techniques in EI and CI ionization in solvent and matrix are presented.

Graphical presentation of the sensitivity variations from pair comparison of the MS modes in EI and CI ionization in solvent and matrix: The first and second MS mode that are compared in each category of the x axis are refered to as A and B. By convention, the terms “increase” refers to enhanced sensitivity of mode A and the term “decrease” refers to enhanced sensitivity of mode B
Comparing full scan and SIM, in CI mode, the majority of the analytes diluted in solvent (95 %) showed higher sensitivity with the full scan technique and only 4 % of the analytes higher sensitivity in SIM. The enhancement (increase) in sensitivity using full scan compared with SIM was over 1,000 % for 89 % of the analytes. As a matter of fact, all organochlorine, organophosphorus and pyrethrinoid pesticides in solvent and in matrix when analyzed with CI–full scan presented 1,000 % enhanced (increased) sensitivity than when analyzed with CI–SIM. This is attributed to the fact that mass spectra produced by CI usually contain a smaller number of fragments [11]. For the analytes that were more sensitive with SIM (4 %), the difference (increase) in sensitivity was between 50 and 100 %. Similar results were observed for the analytes diluted in matrix.
In the EI mode, the analytes which show higher sensitivity in full scan and SIM are approximately 80 and 20 %, respectively. However, for the analytes that present better sensitivity in full scan the enhancement is more than 500 %, while for the analytes with better performance in SIM the same figure lies between 50 and 100 %. At group level, all organochlorines in solvent present more than 1,000 % better sensitivity with full scan mode in comparison with SIM.
Between the full scan and MS/MS in CI mode, the majority of the analytes (93 %) in solvent presented more than 500 % better sensitivity in the full scan mode. The remaining 6 % of analytes exhibited 50–100 % better sensitivity with MS/MS than when analyzed with full scan. The corresponding numbers in matrix are very similar, 91 and 4 %. Organochlorine, organophosphorus and pyrethrinoid pesticides in solvent and in matrix when analyzed with CI–full scan presented an increase of more than 1,000 % in sensitivity in most cases compared to CI–MS/MS. As reported by Alder et al. [11], these three pesticide classes are the ones on which chemical ionization is focused on.
Contrary to CI, in the EI mode, the majority of the analytes (68 % both in solvent and matrix) presented higher sensitivity in the MS/MS mode. The remaining analytes were more sensitive with the full scan technique. However, as seen in Fig. 2, the differences in sensitivity that arise from the comparison of the two techniques are not as intense as in the previous cases. From the experiments, it was made clear that when in matrix, organochlorines have better sensitivity with the MS/MS technique. For organophosporus pesticides, both in solvent and in matrix, no clear differences were detected from the comparison of the two techniques. All pyrethrinoids were more sensitive to MS/MS with difference of up to 100 % when compared with full scan. Flucythrinate is the sole exception, being more sensitive with full scan in matrix.
Comparing the two techniques (SIM and MS/MS) where a pre-selection of ions is required, in CI mode 65 % of the analytes in solvent performed better in SIM. This figure drops slightly when the analytes are in matrix (57 %). All organochlorine analytes are more sensitive to SIM, in contrast to organophoshorus and pyrethrinoids which are spread between SIM and MS/MS equally.
Most notably, in EI mode, all analytes both in solvent and matrix present higher sensitivity in the MS/MS technique with an increase up to 100 % for most of them. This is very well represented in Fig. 2 and 3, where the black columns (rods) have their higher values corresponding to EI–MS/MS in both solvent and matrix. This explains why it is the most used ionization mode for the determination of pesticides in food [1].

Graphical presentation of the sensitivity variations from pair comparison of EI and CI with the three MS techniques, in solvent and matrix. By convention, the term “increase” refers to enhanced sensitivity in CI and the term “decrease” refers to enhanced sensitivity in EI
Comparison of the Two Ionization Modes
For an estimation of the significance of the ionization mode in sensitivity, the two different ionization modes, EI and CI, were compared and the differences were estimated as % increase or decrease of sensitivity. In Fig. 3, a graphical presentation of the sensitivity variations from pair comparison of EI and CI with the three MS techniques in both solvent and matrix are presented.
As obvious from Fig. 3, the sensitivity of the analytes was found to be higher in CI when the analytes were detected using full scan and SIM either in solvent or in matrix. On the contrary, when MS/MS was applied, a significant percentage of the analytes (52 % in solvent and 47 % in matrix) presented higher sensitivity in EI mode. This is in accordance with reported results in literature and is attributed to the fact that mass spectra produced by CI usually contain a smaller number of fragments [11].
At group level, all of the organochlorine pesticides detected with MS/MS presented greater sensitivity in the EI in comparison with CI with an enhancement ranging between 50 and 100 %.
Assessment of Matrix Effect
To estimate the influence of the matrix in sensitivity, two different batches of working solutions were compared and the differences were estimated as % sensitivity increase or decrease. In Fig. 4, a graphical presentation of the sensitivity variations from pair comparison of the solvent and matrix in the two ionization modes and with the three MS techniques are presented.

Graphical presentation of the sensitivity variations from pair comparison of the solvent and matrix in the two ionization modes and with the three MS techniques. By convention, the term “increase” refers to enhanced sensitivity in solvent and the term “decrease” refers to enhanced sensitivity in matrix
In most cases, the sensitivity of the analytes is enhanced when they are diluted in matrix. This is also reported by Hernandez et al. [1]. The sensitivity of the analytes varies depending on the ionization and MS technique. In CI mode, 65 % of the analytes determined with full scan, 89 % with SIM and 86 % with MS/MS exhibit better sensitivity when diluted in matrix. In most cases for full scan and SIM, the difference in sensitivity of the analytes when analyzed in matrix and in solvent is up to 50 % except in the case of MS/MS, in which the difference is up to 100 %. Similar behavior is observed in EI mode, in which 44 % of the analytes determined with full scan, 59 % with SIM and 51 % with MS/MS present higher sensitivity when diluted in matrix. Likewise, the increase is up to 50 % in most cases, except in full scan in which 23 % of the analytes present increase between 50 and 100 %.
At group level, organochlorines present higher sensitivity in matrix in CI mode and higher sensitivity in solvent with EI mode. Pyrethrinoids present higher sensitivity in solvent in both ionization modes with an increase up to 100 %. Organophosphorus presents higher sensitivity in solvent in CI and EI modes with an increase up to 50 % and in CI–MS/MS up to 100 % and higher sensitivity in matrix in EI–SIM with an increase up to 100 %.
Conclusions
Analyses with MS/MS present very good correlation coefficients r for linearity of response both in EI and CI with the analytes in tomato matrix. MS/MS also proved to be more sensitive than SIM in EI mode in both solvent and matrix. In CI mode it was vice versa, with SIM being more sensitive than MS/MS in both solvent and matrix. In comparison to full scan, SIM proved to be more sensitive in CI, but not in EI mode in which the opposite behavior was observed. In addition, full scan proved to be more sensitive than SIM in CI mode, but with the EI mode the variation between the analytes did not lean toward either technique. Comparing ionization modes, CI proved to be more sensitive than EI in all cases. Regarding one of the most crucial analytical problems, in most cases the sensitivity of the analytes is enhanced when diluted in matrix regardless of MS or ionization technique. The findings are in accordance with the reported results in literature.
References
Hernandez F, Cervera MI, Portoles T, Beltran J, Pitarch E (2013) Anal Methods 5:5875–5894
Fernandez-Alba AR, Garcia-Reyes JF (2008) TrAC Trends Anal Chem 27:973–990
Petrovic M, Farre M, de Alda ML, Perez S, Postigo C, Kock M, Radjenovic J, Gros M, Barcelo D (2010) J Chromatogr A 1217:4004–4017
Malik AK, Blasco C, Pico Y (2010) J Chromatogr A 1217:4018–4040
Soler C, Pico Y (2007) TrAC Trends Anal Chem 26:103–115
Lehotay SJ, Mastovska K, Amirav A, Fialkov AB, Martos PA, Kok Ad, Fernandez-Alba AR (2008) TrAC Trends Anal Chem 27:1070–1090
Gomez-Ramos MM, Ferrer C, Malato O, Aguera A, Fernandez-Alba AR (2013) J Chromatogr A 1287:24–37
Pico Y, Blasco C, Font G (2004) Mass Spectrom Rev 23:45–85
Pico Y, Font G, Ruiz MJ, Fernandez M (2006) Mass Spectrom Rev 25:917–960
Barcelo D, Petrovic M (2007) TrAC Trends Anal Chem 26:2–11
Alder L, Greulich K, Kempe G, Vieth B (2006) Mass Spectrom Rev 25:838–865
Ali MA, Baugh PJ (2003) Int J Environ Anal Chem 83:923–933
Huskova R, Matisova E, Svorc L, Mocak J, Kirchner M (2009) J Chromatogr A 1216:4927–4932
Liapis KS, Aplada-Sarlis P, Kyriakidis NV (2003) J Chromatogr A 996:181–187
Raina R, Hall P (2008) Anal Chem Insights 3:111–125
Martinez Vidal JL (2006) Arrebola Liebanas FJ, Gonzalez Rodriguez MJ, Garrido Frenich A, Fernandez Moreno JL. Rapid Commun Mass Spectrom 20:365–375
Tagami T, Kajimura K, Takagi S, Satsuki Y, Nakamura A, Okihashi M, Akutsu K, Obana H, Kitagawa M (2007) Yakugaku Zasshi 127:1167–1171
Turci R, Bruno F, Minoia C (2003) Rapid Commun Mass Spectrom 17:1881–1888
Kawanaka Y, Sakamoto K, Wang N, Yun S-J (2007) J Chromatogr A 1163:312–317
Thompson M, Ellison Stephen LR, Wood R (2002) Harmonized guidelines for single-laboratory validation of methods of analysis (IUPAC Technical Report) Pure Appl Chem, p 835
Method validation and quality control procedures for pesticide residues analysis in food and feed (2011) European Commission, Directorate-General for Health and Consumers (DG SANCO), Brussels, http://ec.europa.eu/food/plant/protection/pesticides/docs/qualcontrol_en.pdf. Accessed 18 June 2014
Anastassiades M, Lehotay SJ, Tajnbaher D, Schenck FJ (2003) J AOAC Int 86:412–431
Miller JN, Miller CN (2010) Statistics and Chemometrics for Analytical Chemistry. Pearson Education Limited, Edinburgh
Trufelli H, Palma P, Famiglini G, Cappiello A (2011) Mass Spectrom Rev 30:491–509
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Anagnostopoulos, C., Charalampous, A. & Balayiannis, G. EI and NCI GC–MS and GC–MS/MS: Comparative Study of Performance Characteristics for the Determination of Pesticide Residues in Plant Matrix. Chromatographia 78, 109–118 (2015). https://doi.org/10.1007/s10337-014-2800-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10337-014-2800-z