
Abstract
Blood samples from 94 coal tits (Parus ater), 56 great tits (Parus major) and 219 pied flycatchers (Ficedula hypoleuca), caught between 1993 and 2002 at two localities in Lower Saxony, Germany, were examined for haemosporidian infection by parasite-specific polymerase chain reaction (PCR). A simple PCR targeting the 18 SSU rRNA gene of the parasites was used for rapid screening of the samples and generated a total infection prevalence of 20.6% (76/369): 6.8% (n = 15) of the pied flycatchers, 19.1% (n = 18) of the coal tits and 76.8% (n = 43) of the great tits were infected. The positive specimens were re-examined by a cytochrome b gene-directed nested PCR producing significantly longer DNA fragments (approx. 520 bp) that were sequenced and analysed against GenBank-deposited nucleotide sequences. In various numbers (once to 30 times), a total of 13 parasitic DNA sequences differing from 2.9 to 8.5% (13–45 nucleotides) were demonstrated in the three bird species. Due to similarities of 98–100% with GenBank entries, 11 sequences could be assigned to Plasmodium sp. and two to the genus Haemoproteus. In summary, 57 birds were infected with Plasmodium and 19 with Haemoproteus, corresponding to 15.4 and 5.1% of all birds examined, and to 75 and 25% of all birds tested positive. As the only defined species, Haemoproteus majoris was identified in 17 great tits.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
With few exceptions, avian haemosporidia occur worldwide, irrespective of climatic barriers which limit the distribution of human haemosporidia, i.e. Plasmodium parasites that cause malaria (Smyth 1976; Seed and Manwell 1977). According to Manwell (1935), the vast distribution of haemosporidian infection in birds is due to the high mobility and migration pattern of the host species. Additionally, there is limited host specificity on the vector-side, i.e. in the culicid mosquitoes, ceratopogonid midges, simuliid and hippoboscid flies (Olsen 1974; Rathore et al. 2001; Valkiūnas 2005) transmitting the various parasite genera.
The haemosporidian species parasitising birds belong to the genera Plasmodium, Haemoproteus and Leucocytozoon (Valkiūnas 2005). At present, there are 34 valid taxonomic species of the genus Plasmodium, 134 of the genus Haemoproteus and 60 of the genus Leucocytozoon (Bennett et al. 1993, 1994). While most Leucocytozoon species are relatively host-specific and restricted to closely related species within the same host family (Fallis et al. 1974), Haemoproteus and Plasmodium parasites appear to exhibit a low degree of host specificity and occur in several bird families (Bishop and Bennett 1992; Szymanski and Lovette 2005).
Despite our knowledge on the geographic distribution of avian haemosporidia in general, there are only few and fragmentary data on their ecologies, their infection epidemiologies and their prevalences with respect to host species. Depending on the geographical region and bird species examined, infection prevalences range from 0.6 to 19% for Plasmodium, from 2.6 to 19.5% for Haemoproteus, and from 0.1 to 17.7% for Leucozytozoon parasites (Greiner et al. 1975; Smyth 1976; McClure et al. 1978; Gabaldon et al. 1974, 1975, 1976). For Europe, Kučera (1981) found 11.3% out of 10,194 birds infected with Plasmodium, 11.8% with Haemoproteus and 5.9% with Leucocytozoon species, while Rintamäki et al. (1997, 2000) calculated infection prevalences of 0.8% for Plasmodium, 4.2% for Haemoproteus, and 10.1% for Leucocytozoon, on a basis of 308 birds examined.
All the data previously mentioned were obtained by examination of avian blood smears by light microscopy. However, contrary to other haemosporidian parasites, including Haemoproteus and Leucocytozoon species, avian plasmodia can re-invade various tissues where they cannot be detected by light microscopy following their red blood cell passage. Thus, in bird species adapted to the parasites, the parasitemia soon declines after an initial increase, and eventually the parasite may even disappear from the blood. Nevertheless, the bird remains infected with dormant tissue stages and may be a possible source of vector infection in the case of a relapse (Waldenström et al. 2004). Due to the primary tissue confinement of the parasites and the limited sensitivity of light microscopy, the actual infection prevalences are usually considerably underestimated in epidemiological studies (Valkiūnas and Iezhova 2001).
While an infection with haemosporidian parasites generally does not cause serious harm to a bird as long as mutual adaptation has been acquired by coevolution (Garnham 1966), the accidental introduction of allochthonous parasite strains can lead to high morbidity and mortality rates among the indigenous bird fauna (Bensch et al. 2000), and even result in endangering endemic bird species as may presently be observed on Hawaii (Massey et al. 1996; Woodworth et al. 2005). The reverse situation, where imported birds come into contact with indigenous haemosporidian parasites, is possible as well, and can lead to enormous economic loss in zoological gardens or private livestock husbandry when highly susceptible exotic birds such as penguins are infected (Lindt and Hörning 1966; Kronberger and Schüppel 1977; Valentin et al. 1994).
Further knowledge on the epidemiology and pathology of haemosporidian infection in birds is therefore of major importance. The presented study was meant to contribute data to the occurrence of avian haemosporidia in Germany by determining infection prevalences and the pathogen spectrum in Lower Saxonian bird populations by means of molecular biological techniques.
Methods
Origin of samples
All samples analysed originally stem from work on the occurrence of extra-pair paternity in three bird species (e.g. Brün et al. 1996; Lubjuhn et al. 1999a, b, 2000; Schmoll et al. 2003), coal tits (Parus ater, Fam. Paridae), great tits (Parus major, Fam. Paridae), and pied flycatchers (Ficedula hypoleuca, Fam. Muscicapidae). From these large collections, random subsamples of blood specimens that all stem from adult birds were examined: 94 coal tits, 56 great tits and 219 pied flycatchers. The respective birds had been trapped in their nest boxes while feeding nestlings from May to June of the years 1993, 1996, 1999 and 2002 in the boroughs of Lingen (52°27′N, 7°15′E) and Bahrdorf (52°23′N, 10°59′E) in Lower Saxony, Germany. Blood samples had been taken by puncturing the ulnar vein with a canula and recovering the leaking blood (about 50 μl) by a glass capillary (please note that this sort of blood sampling has no detectable negative effects on the birds; see Lubjuhn et al. 1998; Schmoll et al. 2004). The blood samples were transferred to 250 μl APS-buffer (Arctander 1988) and stored at –20°C until processing. DNA was extracted according to a modified standard protocol (Miller et al. 1988; Lubjuhn and Sauer 1999) yielding about 200 μl of DNA solution.
DNA amplification
To begin with, each DNA sample was screened for haemosporidian parasites by a polymerase chain reaction (PCR) specifically amplifying a 243 bp fragment of the parasitic 18 SSU rRNA-gene (Hulier et al. 1996). Amplification was performed in a total of 50 μl containing 10 mM Tris–HCl, pH 8.3, 50 mM KCl, 200 nM dNTPs, 120 nM of primers rPLU3 and rPLU4, 6 mM MgCl2, 1 U Taq DNA-polymerase (Invitrogen) and 10 μl of DNA-solution. Thermoconditions were 5 min at 95°C initially, followed by 35 cycles of 2 min at 95°C (denaturation), 2 min at 64°C (primer annealing), and 1 min at 72°C (DNA extension), and finalised by 10 min at 72°C.
In a second step, the DNA samples tested positive were examined by a further PCR targeting a parasitic cytochrome b (cyt b) gene region of about 520 bp (Waldenström et al. 2004) to produce a DNA-fragment of sufficient length and sequence information to be analysed in comparison with GenBank derived nucleotide sequences of haemosporidian parasites. Besides the length argument, there are much more cyt b than 18 SSU rRNA gene sequence entries in the GenBank available. The nested PCR according to Waldenström et al. (2004) was done in a volume of 25 μl using primers HAEMNF and HAEMNR2 in the first, and primers HAEMF and HAEMR2 in the second round of amplification. The composition of reaction mixtures were the same for both PCRs: 10 mM Tris–HCl, pH 8.3, 50 mM KCl, 400 nM dNTPs, 600 nM of each of the two primers, 1.5 mM MgCl2, 1.25 U Taq DNA-Polymerase (Invitrogen), and 1 μl of DNA-solution. The thermoprofiles consisted of an initial 3 min at 94°C, 20 (1. nested) and 35 cycles (2. nested), respectively, of 30 s at 94°C, 30 s at 50°C, and 45 s at 72°C, and a final 10 min at 72°C.
In some cases, when no amplification product was obtained, the volume of template DNA was increased stepwise up to 10 μl for the first and up to 5 μl for the second round of nested PCR. DNA that still produced no PCR products was unspecifically preamplified using the ‘GenomiPhi Kit’ (Amersham Biosciences) prior to processing by the specific nested PCR. PCR products were run on 1.5% agarose gels at 120 V for approx. 1 h and, after ethidiumbromide-staining (0.5 μg/ml), visualised by UV light (312 nm).
DNA sequencing
After electrophoresis, the nested PCR products were excised from the gels and eluted by the ‘QIAamp Gel Extraction Kit’ (Qiagen) according to the manufacturer’s instructions. The eluted DNA was again electrophoresed to check for quality and quantity, and 20–30 ng per 100 bp of fragment length were prepared for sequencing. Sequencing was done in both directions in duplicate by cycle sequencing on an ABI Prism 377 DNA sequencer (Applied Biosystems) using one of the PCR primers as sequencing primer.
Sequence analysis
Cyt b sequences obtained were blasted (www.ncbi.nlm.nih.gov/BLAST/blastn) with all equivalent sequence entries for haemosporidia available in the GenBank. They were assigned to species or genus linked to those GenBank sequences with highest percentages of concordance.
Statistical analysis
Statistical analysis of the birds’ infection prevalences was performed with the computer program SPSS 10.0 applying the Gauss test (G test). Probability values of P ≤ 0.05 were considered significant.
Results
Of the 369 avian blood samples collected, 76 were tested positive for haemosporidian parasites (Table 1) by specific PCR-amplification of their 18 SSU rDNA. The overall infection prevalence for the birds from Bahrdorf was more than four times higher than that for the birds from Lingen (G test: G = 44.6, P < 0.001, df = 1). However, it has to be kept in mind that no great tits from Lingen were available for examination, as these contribute significantly to the uneven ratio between the two localities in that more than three-quarters of them were tested positive. Coal tits were by far more often infected in Bahrdorf than in Lingen (G test: G = 5.7, P < 0.028, df = 1), whereas the pied flycatchers had nearly the same, relatively low infection prevalences at both localities (G test: G = 0.002, P = 0.97, df = 1). However, in these instances it has to be noted that the subsamples do not only stem from different localities, but also from different years.
In summary, great tits were most often infected, followed by coal tits and pied flycatchers (G test; great tits/coal tits: G = 12.3, P < 0.001, df = 1; great tits/pied flycatchers: G = 72.4, P < 0.001, df = 1; coal tits/pied flycatchers: G = 11.1, P = 0.001, df = 1). In none of the bird species, significant differences in infection prevalences according to sex were recorded (data not shown).
DNA sequencing of the parasitic cyt b gene nested PCR products of the 76 positive samples resulted in a total of 13 different DNA sequences (Table 2) varying in length from 522 to 528 nucleotides. Variations amounted to 2.9–8.5%, i.e. 13–45 nucleotides. In 59 birds, DNA sequences were demonstrated that had complete identity with sequences deposited in the GenBank (see asterisks in Table 2). The nucleotide sequences of the other 17 samples still had high similarities of more than 98% with GenBank entries. One sequence (identical to GenBank entry AF495571 for Plasmodium sp.) was found 30 times in all three bird species and at both localities. Another sequence occurring in 17 great tit blood samples was 100% homologous to the GenBank entry AF254977 for Haemoproteus majoris. At the same time, this was the only DNA sequence that could be assigned to a defined parasite species, whereas the other sequences, although in many cases identical to GenBank entries, could only be assigned to genus level due to a lack of species information. A third DNA sequence with high similarity to GenBank entry AF069611 for Plasmodium sp. was represented 11 times in coal and great tits, and a fourth sequence with high homology to GenBank entry AF495576 for Plasmodium sp. was found five times in coal tits and pied flycatchers. The remaining nine sequences were detected once in three birds, twice in two birds each, and six times in one single bird each, but each of them in one bird species only. Double-infections were not observed as sequencing chromatograms displayed no overlapping peaks in the otherwise clean reactions.
After analysis of the DNA sequences, it was possible to calculate the infection prevalences according to parasite genus and host species. Of the 369 birds examined, 57 were parasitised by Plasmodium sp. (15.4%) and 19 by Haemoproteus sp. (5.1%), corresponding to 75 and 25%, respectively, of the 76 birds found infected. Itemised to the three bird species, infection prevalences were: 30.4% (n = 17) Haemoproteus sp. and 46.4% (n = 26) Plasmodium sp. in great tits, no Haemoproteus sp. and 18.7% (n = 18) Plasmodium sp. in coal tits, and 0.9% (n = 2) Haemoproteus sp. and 5.9% (n = 13) Plasmodium sp. in pied flycatchers.
Although only three bird species were involved in this study, we found two Haemoproteus and 11 Plasmodium strains/species. Both Haemoproteus and eight Plasmodium lineages were demonstrated in one single host species each. The pied flycatcher harboured one Haemoproteus and nine Plasmodium lineages. Only three parasite lineages (all of them Plasmodium sp.) were present in more than one host species. Certain Plasmodium lineages could be found in different years of the study, but only one of them in the same host species in different study years. The most prevalent Plasmodium lineage (AF495571) was demonstrated in all four study years.
Discussion
There are few studies on the occurrence and distribution of avian infection with haemosporidian parasites in Germany. Krone et al. (2001), for example, examined 1,149 owls and birds of prey and found a total of 11% of the birds infected, including 110 Leucocytozoon cases (9.6%), 37 Haemoproteus cases (3.2%), and one single bird harbouring plasmodia (0.1%). Haberkorn (1984) determined a similar total infection prevalence of 14% in 893 passerines from 44 species and 14 families: 6.4% of the birds were infected with Leucocytozoon, 4.6% with Haemoproteus and 1.1% with Plasmodium species. Among the birds examined were 82 P. major of which six (7.3%) were infected with Haemoproteus and one (1.2%) with Leucocytozoon. Four P. ater specimens were found not to be parasitised. Kronberger and Schüppel (1977) published a study on 7,126 autopsies on birds from German zoological gardens according to which 0.3% (22 cases) of Leucocytozoon infections, mainly in parrots, and 0.2% (14 cases) of plasmodial infections, mainly in penguins, were diagnosed.
While the total infection prevalences obtained in our study were comparable to previous studies carried out in Germany for Haemoproteus, they were much higher with regard to Plasmodium. This may be partly due to the detection technique applied (PCR versus light microscopy), provided that the PCR does not detect Haemoproteus as efficiently as Plasmodium. Actually, the PCR method according to Hulier et al. (1996), which was used for blood screening, was originally described for the specific amplification of Plasmodium-DNA and, prior to our experiments, we did not expect it to detect Haemoproteus or Leucocytozoon. While Leucocytozoon-DNA was indeed not amplified, Haemoproteus was regularly demonstrated in the samples. It remains, however, to be elucidated whether all species of this systematic group can be detected by this PCR approach.
Unfortunately, avian blood smears for light microscopical examination to better compare our results with former results were not available. While PCR is supposed to be more sensitive than light microscopy, it may produce false positive results as a consequence of sample contamination. This, however, can virtually be excluded for this study as we have followed the usual precautionary measures and, moreover, could consistently amplify parasitic DNA from each of the 76 positive blood samples with two completely different PCR approaches.
As was also unknown prior to our experiments, the simple PCR according to Hulier et al. (1996), which targets ribosomal DNA, is obviously more efficient in demonstrating a parasite infection than the nested PCR according to Waldenström et al. (2004), which targets mitochondrial DNA, as can be deduced from the increase and preamplification of DNA necessary in the case of the nested PCR.
The infection prevalences obtained display huge and, in the majority, significant differences between the bird species involved. Reasons can be manifold and related to parasite host, vector, locality and/or time (year) of sampling.
Thus, for example, a different basic susceptibility for the parasites and behavioural aspects of the three bird species may contribute to the observed differences in the infection prevalences. Parasite transmission may be influenced by the size of the breeding territory (distance between hatcheries) as well as by the pairing and flocking behaviour. The high number of different parasite sequences found in pied flycatchers (10 different sequences in 15 infected birds, compared to only 3 sequences in 43 infected great tits and 4 in 18 infected coal tits, respectively; see also Table 2) may, at least partially, be explained by the hosts’ migratory behaviour. Due to their seasonal travels between Europe and Africa, pied flycatchers are presumably exposed to many more geographical strains and/or species of parasites than are non-migratory birds such as the Parus species investigated.
At first sight, infection prevalences in pied flycatchers are surprisingly low in view of the comparatively high number of different parasite sequences found. But exposure to many different strains or species may create high selective pressures which in turn may lead to adaptations that help to circumvent and/or deal with an actual infection. It has to be kept in mind, however, that blood samples did not only stem from two different study sites, but, at least partly, from different years with possible between-year differences. Avoidance of overvaluation of such natural phenomena can only be achieved by longitudinal studies.
With respect to the localities, differences in the vector fauna responsible for parasite transmission are conceivable. Both the abundance and the differential vector competence of the species occurring may play a role. Differing types of forests and regional or local micro-climatic factors may influence the vector fauna and, ultimately, the birds’ infection prevalences.
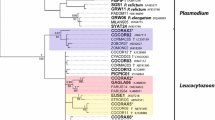
Species identification and systematics in general is a profound problem in avian haemosporidia, particularly within the genus Plasmodium (Waldenström et al. 2004), which is partly reflected by the ongoing debate on the use of the term ‘malaria parasite’ (Pérez-Tris et al. 2005; Valkiūnas et al. 2005). Classical systematics, which is based on conventional, mainly morphological criteria, distinguishes four Plasmodium subgenera (Corradetti et al. 1963). However, recent molecular biological studies do not support the species relationships set by this classification, but argue for other ones (Kissinger et al. 2002). Moreover, as other studies on parasitic genes PCR-amplified from avian hosts have shown before (e.g. Perkins and Schall 2002; Ricklefs and Fallon 2002), there are numerous variations in nucleotide sequence which can, in most cases, not be assigned to a known and morphologically defined strain or species. This is the reason why there are various GenBank sequence entries for haemosporidian parasites only denoted by its genus. Actually, some scientists are of the opinion that every unique DNA sequence marks a separate parasite species (Bensch et al. 2004) and, according to Perkins (2000), sequence variations of at least 3% should be defined as separate species.
In the study presented, we were also confronted with the problem of sequence polymorphism when comparing the cyt b gene sequences of the haemosporidia detected in the three bird species with GenBank-deposited sequences. Although parasites from 59 birds had gene sequences identical to GenBank ones, most of them could only be identified to genus but not to species level. Only one single sequence detected in 17 great tits from Bahrdorf was 100% homologous to a GenBank sequence linked to a species name, namely H. majoris. Seventeen bird parasite sequences had no identical counterparts in the GenBank and thus were assigned to a genus by the highest possible sequence similarity. By doing so, we found 75% of all sequences characteristic for Plasmodium and 25% of the sequences characteristic for Haemoproteus species. In detail, 11 different sequences were assigned to the genus Plasmodium and two sequences to the genus Haemoproteus. Of these 13 haplotypes, ten occurred in F. hypoleuca, four in P. major, and three in P. ater. The same number of Haemoproteus and Plasmodium haplotypes was demonstrated by Bensch et al. (2000) in a study on 278 passerine birds from 12 species collected in Europe, Africa and Asia, whereby only one of the haplotypes was found in more than one host species, namely H. majoris in three great tit specimens and in four blue tit (P. caeruleus) specimens. While we found H. majoris only in great tits, Bishop and Bennett (1992) list this parasite species to occur in European coal tits as well. They mention additional unknown Haemoproteus lineages in various other bird species, including the ones found in this study. According to their compilation, various tit species, including great tits, were found infected with several Plasmodium strains/species, among them P. relictum, P. polare, P. circumflexum and P. vaughani, whereas in coal tits, no plasmodia were described at all. In pied flycatchers, only P. vaughani and some unknown Plasmodium lineages were demonstrated. Thus, our findings correspond mainly to Bishop and Bennett’s compendium, except that we could identify several Plasmodium lineages in coal tits.
Due to the limited information about the parasite lineages found in this study it was not possible to analyse them with regard to other parameters, such as their hosts’ and vectors’ ecology, interspecific interactions, coevolution or pathology. Sequencing and matching the parasite strains with GenBank data is, however, an extremely powerful tool, and collecting more and more strain information will one day allow producing differential prevalence patterns in bird populations.
Our study suggests that avian infection with haemosporidian parasites is much more frequent in Germany than previous data have indicated, and that numerous parasite strains and/or species circulate. When considering such infection prevalences obtained by PCR, it must, however, be kept in mind that a certain proportion of the birds might be dead-end hosts for some parasite strains, which in that case will not be able to develop in the birds, but still be detectable in the blood for some time by amplification of their DNA by the sensitive PCR method. On the other hand, a high prevalence of active stages of parasites—at least of endemic species—in the birds’ blood is not principally surprising during the avian breeding season as the vector abundance is supposed to be high at this time of the year and the hosts’ immune system is challenged by reproductive effort. Nevertheless, the data obtained may probably still underestimate reality as further birds must be supposed to be infected with parasitic tissue stages only, not detectable in living birds by blood examination. Moreover, the PCR systems used were most likely not able to detect species of the third haemosporidian genus occurring in birds, Leucocytozoon. This deficit can be overcome in future research by applying a newly developed PCR approach (Hellgren et al. 2004).
Zusammenfassung
Infektion mit Haemosporidia bei Singvögeln aus Niedersachsen
Mittels einer Parasiten-spezifischen Polymerase-Kettenreaktion (PCR) wurden Blutproben von 94 Tannenmeisen (P. ater), 56 Kohlmeisen (P. major) und 219 Trauerschnäppern (F. hypoleuca), die zwischen 1993 und 2002 in Untersuchungsgebieten in der Nähe der niedersächsischen Gemeinden Bahrdorf und Lingen gesammelt worden waren, auf eine Infektion mit Haemosporidia untersucht. Ein schnelles Screening der Proben mit einer einfachen PCR, welche das 18 SSU rRNA-Gen der Parasiten zur Zielsequenz hatte, erbrachte eine gesamte Infektionsprävalenz von 20,6% (76/369): 6,8% (n = 15) der Trauerschnäpper, 19,1% (n = 18) der Tannenmeisen und 76,8% (n = 43) der Kohlmeisen waren infiziert. Die positiven Proben wurden im Folgenden einer Cytochrom b-Gen spezifischen nested-PCR unterzogen, welche bedeutend längere DNA-Fragmente (ca. 520 bp) liefert. Diese wurden sequenziert und mit in der GenBank abgelegten DNA-Sequenzen verglichen. Insgesamt wurden 13 verschiedene DNA-Sequenzen ermittelt, die bei den drei untersuchten Vogelarten unterschiedlich häufig vorkamen (ein- bis 30mal) und sich in ihrer Sequenz zwischen 2,9 und 8,5% (13 bis 45 Nukleotide) voneinander unterschieden. Aufgrund von Sequenzübereinstimmungen zwischen 98 und 100% mit den in der GenBank hinterlegten Einträgen konnten 11 Sequenzen der Gattung Plasmodium und zwei Sequenzen der Gattung Haemoproteus zugewiesen werden. Insgesamt waren 57 Vögel mit Plasmodium infiziert und 19 mit Haemoproteus, entsprechend 15,4 bzw. 5,1% aller untersuchter Vögel und 75 bzw. 25% aller positiver Vögel. Als einzige definierte Erregerspezies konnte Haemoproteus majoris bei 17 Kohlmeisen identifiziert werden.
References
Arctander P (1988) Comparative studies on avian DNA restriction fragment length polymorphism analysis: convenient procedures based on blood samples from live birds. J Ornithol 129:205–216
Bennett GF, Bishop MA, Peirce MA (1993) Checklist for the avian species of Plasmodium Marchiafava & Celli, 1885 (Apicomplexa) and their distribution by avian family and Wallacean life zones. Syst Parasitol 26:171–179
Bennett GF, Peirce MA, Earlé RA (1994) An annotated checklist of the valid avian species of Haemoproteus, Leucocytozoon (Apicomplexa: Haemosporida) and Hepatozoon (Apicomplexa: Haemogregarinidae). Syst Parasitol 29:61–73
Bensch S, Stjernman M, Hasselquist D, Östman Ö, Hansson B, Westerdahl H, Pinheiro RT (2000) Host specificity in avian blood parasites: a study of Plasmodium and Haemoproteus mitochondrial DNA amplified from birds. Proc R Soc Lond B 267:1583–1589
Bensch S, Perez-Tris J, Waldenström J, Hellgren O (2004) Linkage between nuclear and mitochondrial DNA sequences in avian malaria parasites: multiple cases of cryptic speciation. Evolution 58:1617–1621
Bishop MA, Bennett GF (1992) Host–parasite catalogue of the avian haematozoa, supplement 1. Occas Pap Biol (Mem Univ Newfoundland) 15:1–211
Brün J, Winkel W, Epplen JT, Lubjuhn T (1996) Elternschaftsnachweise bei Trauerschnäppern Ficedula hypoleuca am Westrand ihres mitteleuropäischen Verbreitungsareals. J Ornithol 137:435–446
Corradetti A, Garnham PCC, Laird M (1963) New classification of the avian malaria parasites. Parassitologia 5:1–4
Fallis AM, Desser SS, Khan RA (1974) On species of Leucocytozoon. Adv Parasitol 12:1–67
Gabaldon A, Ulloa G, de Montcourt AG (1974) Encuesta sobre malaria aviara en Venezuela: resultados del primer año. Bol Direc Malariol Saneam Ambient 14:80–104
Gabaldon A, Ulloa G, de Montcourt AG (1975) Encuesta sobre malaria aviara en Venezuela: resultados del segundo año. Bol Direc Malariol Saneam Ambient 15:73–92
Gabaldon A, Ulloa G, de Montcourt AG (1976) Encuesta sobre malaria aviara en Venezuela: resultados del tercer y último año. Bol Direc Malariol Saneam Ambient 16:107–118
Garnham PCC (1966) Malaria parasites and other haemosporidia. Blackwell, Oxford
Greiner EC, Bennett GF, White EM, Coombs RF (1975) Distribution of the avian hematozoa of North America. Can J Zool 53:1762–1787
Haberkorn A (1984) Observations on malaria in European perching birds (Passeriformes). Zbl Bakt Hyg A 256:288–295
Hellgren O, Waldenström J, Bensch S (2004) A new PCR assay for simultaneous studies of Leucocytozoon, Plasmodium, and Haemoproteus from avian blood. J Parasitol 90:797–802
Hulier E, Petour P, Snounou G, Nivez M-P, Miltgen F, Mazier D, Renia L (1996) A method for the quantitative assessment of malaria parasite development in organs of the mammalian host. Mol Biochem Parasitol 77:127–135
Kissinger JC, Souza PC, Soares CO, Paul R, Wahl AM, Rathore D, McCutchan TF, Krettli AU (2002) Molecular phylogenetic analysis of the avian malarial parasite Plasmodium (Novyella) juxtanucleare. J Parasitol 88:769–773
Kronberger H, Schüppel K-F (1977) Zwanzig Jahre postmortale Untersuchungen von Vögeln. Verhandlungsber Erkr Zootiere 19:153–169
Krone O, Priemer J, Streich J, Sömmer J, Langgemach T, Lessow O (2001) Haemosporida of birds of prey and owls from Germany. Acta Protozool 40:281–289
Kučera J (1981) Blood parasites of central Europe. 1. Survey of literature. The incidence in domestic birds and general remarks to the incidence in wild birds. Folia Parasitol 28:13–22
Lindt S, Hörning B (1966) Über Malaria und Pinguine. Verhandlungsber Erkr Zootiere 8:223–231
Lubjuhn T, Sauer KP (1999) DNA fingerprinting and profiling in behavioural ecology. In: Epplen JT, Lubjuhn T (eds) DNA profiling and DNA fingerprinting. Birkhäuser, Basel, pp 39–52
Lubjuhn T, Brün J, Winkel W, Muth S (1998) Effects of blood sampling in great tits. J Field Ornithol 69:595–602
Lubjuhn T, Gerken T, Brün J, Epplen JT (1999a) High frequency of extra-pair paternity in the coal tit. J Avian Biol 30:229–233
Lubjuhn T, Strohbach S, Brün J, Gerken T, Epplen JT (1999b) Extra-pair paternity in great tits (Parus major)—a long term study. Behaviour 136:1157–1172
Lubjuhn T, Winkel W, Epplen JT, Brün J (2000) Reproductive success of monogamous and polygynous pied flycatchers (Ficedula hypoleuca). Behav Ecol Sociobiol 48:12–17
Manwell RD (1935) How many species of avian malaria parasites are there? Am J Trop Med 15:265–282
Massey JG, Graczyk TK, Cranfield MR (1996) Characteristics of naturally acquired Plasmodium relictum capistranoae infections in naïve Hawaiian crows (Corvus hawaiiensis) in Hawaii. J Parasitol 82:182–185
McClure HE, Poonswad P, Greiner EC, Laird M (1978) Haematozoa in the birds of eastern and southern Asia. Mem Univ Newfoundland, St. John’s, Newfoundland
Miller SA, Dykes DD, Polesky HF (1988) A simple salting procedure for extracting DNA from human nucleated cells. Nucleic Acids Res 16:1215
Olsen OW (1974) Animal parasites, their life cycles and ecology. University Park Press, Baltimore
Perkins SL (2000) Species concepts and malaria parasites: detecting a cryptic species of Plasmodium. Proc R Soc Lond B 267:2345–2350
Perkins SL, Schall JJ (2002) A molecular phylogeny of malarial parasites recovered from cytochrome b gene sequences. J Parasitol 88:972–978
Pérez-Tris J, Hasselquist D, Hellgren O, Krizanauskiene A, Waldenström J, Bensch S (2005) What are malaria parasites? Trends Parasitol 21:209–211
Rathore D, Wahl AM, Sullivan M, McCutchan TF (2001) A phylogenetic comparison of gene trees constructed from plastid, mitochondrial and genomic DNA of Plasmodium species. Mol Biochem Parasitol 114:89–94
Ricklefs RE, Fallon SM (2002) Diversification and host switching in avian malaria parasites. Proc R Soc Lond B 269:885–892
Rintamäki PT, Halonen M, Kilpimaa J, Lundberg A. (1997) Blood parasites found in three passerine species during spring migration. Ornis Fenn 74:195–200
Rintamäki PT, Ojanen M, Pakkala H, Tynjälä M, Lundberg A (2000) Blood parasites of juvenile willow tits Parus montanus during autumn migration in northern Finland. Ornis Fenn 77:83–87
Schmoll T, Dietrich V, Winkel W, Epplen JT, Lubjuhn T (2003) Longterm fitness consequences of female extra-pair matings in a socially monogamous passerine. Proc R Soc Lond B 270:259–264
Schmoll T, Dietrich V, Winkel W, Lubjuhn T (2004) Blood sampling does not affect fledging success and fledgling local recruitment in coal tits (Parus ater). J Ornithol 145:79–80
Seed TM, Manwell RD (1977) Plasmodia of birds. In: Kreier JP (ed) Parasitic protozoa, vol 3. Academic, New York, pp 311–357
Smyth JD (1976) Introduction to animal parasitology, 2nd edn. Halsted Press, Wiley, New York, pp 112–121
Szymanski MM, Lovette IJ (2005) High lineage diversity and host sharing of malarial parasites in a local avian assemblage. J Parasitol 91:768–774
Valentin A, Haberkorn A, Hensch B, Jakob W (1994) Massive Malaria-Infektionen mit Parahaemoproteus spec. in Schnee-eulen (Nyctea scandiaca) und deren Behandlung mit Primaquin. Verhandlungsber Erkr Zootiere 36:401–404
Valkiūnas G (2005) Avian malaria parasites and other haemosporidia. CRC Press, Boca Raton
Valkiūnas G, Iezhova TA (2001) A comparison of the blood parasites in three subspecies of the yellow wagtail Motacilla flava. J Parasitol 87:930–934
Valkiūnas G, Anwar AM, Atkinson CT, Greiner EC, Paperna I, Peirce MA (2005) What distinguishes malaria parasites from other pigmented haemosporidians? Trends Parasitol 21:357–358
Waldenström J, Bensch S, Hasselquist D, Östman Ö (2004) A new nested polymerase chain reaction method very efficient in detecting Plasmodium and Haemoproteus infections from avian blood. J Parasitol 90:191–194
Woodworth BL, Atkinson CT, LaPointe DA, Hart PJ, Spiegell CS, Tweed EJ, Hennemann C, LeBrun J, Denette T, de Mots R, Kozar KL, Triglia D, Lease D, Gregor A, Smith T, Duffy D (2005) Host population persistence in the face of introduced vector-borne diseases: Hawaii amakihi and avian malaria. Proc Natl Acad Sci USA 102:1531–1536
Acknowledgements
We thank all people who participated in collecting avian blood samples (they are acknowledged in the publications cited in the respective part of the Methods section) and S. Bleidissel, M. Orland and C. Wallnisch (Institute for Evolutionary Biology and Ecology, University of Bonn) for technical laboratory assistance. The studies for which the blood samples were originally obtained were supported by the Deutsche Forschungsgemeinschaft (Lu572/1 and Lu572/2), while the specific aims of this study were supported by the Deutsche Ornithologen-Gesellschaft.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by F. Bairlein.
Rights and permissions
About this article
Cite this article
Wiersch, S.C., Lubjuhn, T., Maier, W.A. et al. Haemosporidian infection in passerine birds from Lower Saxony. J Ornithol 148, 17–24 (2007). https://doi.org/10.1007/s10336-006-0094-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10336-006-0094-0