
Abstract
To improve the diagnosis of Stewart’s wilt, we developed a loop-mediated isothermal amplification (LAMP) assay, based on two conserved sequences of the cpsD and pstS–glmS gene regions. The detection limit of the LAMP assay was 104 colony-forming units/mL. No cross reaction was observed with other Pantoea spp., other genera associated with sweet corn diseases, or strains isolated from the surface of maize leaves in Japan. The LAMP reaction was not inhibited by leaf contents. The simplicity of sample preparation and short processing time make this LAMP assay useful for field surveillance of Stewart’s wilt.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Stewart’s wilt, caused by Pantoea stewartii subsp. stewartii (syn. Erwinia stewartii [Mergaert et al. 1993], hereafter abbreviated Pnss), is one of the most important diseases of sweet corn (Zea mays L. var. rugosa) and inbred field corn. Young seedlings infected with Pnss become severely wilted and die in most cases. The organism is transmitted by the corn flea beetle (Chaetocnema pulicaria). Seed transmission is also possible (Pataky and Ikin 2003). Pnss is endemic throughout a large portion of the maize-growing regions of the eastern and midwestern United States, and it occurs intermittently in Canada.
More than 60 countries have placed quarantine regulations on maize seed produced in Pnss-affected regions to prevent the introduction of the pest (Pataky and Ikin 2003). Because Pnss is technically difficult to detect from seed at a port-of-entry inspection but can be easily detected in the field during the growing season, importation of maize seeds is approved under the condition that the plants concerned are inspected at the growing site in exporting countries (MAFF Plant Protection Station 2014). Therefore, also in noninfested countries and areas, including Japan, field surveillance is essential to eliminate the disease at an early stage. To implement field surveillance effectively, proper and rapid methods are needed to detect Stewart’s wilt from questionable plant materials in the fields.
Several methods to detect and identify P. stewartii have been reported (Coplin et al. 2002; Lamka et al. 1991; Tambong et al. 2008; Wensing et al. 2010), including subspecies-specific methods (Gehring et al. 2014; Xu et al. 2010). Some conventional techniques, such as enzyme-linked immunosorbent assay (Lamka et al. 1991), are time-consuming and relatively insensitive compared with nucleic acid-based methods. Molecular techniques include conventional PCR (Coplin et al. 2002; Gehring et al. 2014; Wensing et al. 2010), ‘miniprimer’ PCR (Xu et al. 2010) and a specific real-time PCR assay (Tambong et al. 2008; Wensing et al. 2010). Some of these methods use the cpsD gene region and the region between the pstS and glmS genes. The cpsD region, part of the cps gene cluster, is required for the production of the exopolysaccharide stewartan, and plays an important role in pathogenicity and virulence (Coplin and Majerczak 1990). The region between pstS and glmS seems to be specific to this species (Wensing et al. 2010). However, while PCR-based methods are rapid, specific and highly sensitive, their practical use is hindered by the need for complex and expensive thermal cycling equipment and time-consuming processes. Inhibition of PCR by contaminant plant components also makes it difficult to detect Pnss in plant materials.
Loop-mediated isothermal amplification (LAMP) is a new molecular diagnostic technique that is simple, rapid and sensitive (Notomi et al. 2000). It does not require expensive thermal cycling equipment because DNA amplification uses only one enzyme, Bst DNA polymerase, under an isothermal condition of 60° to 65 °C (Notomi et al. 2000). The LAMP reaction uses a set of four specially designed primers (FIP, BIP, F3, B3) that recognize six distinct sequences on the target (Notomi et al. 2000). The reaction is accelerated by the addition of loop primers (Nagamine et al. 2002). The LAMP reaction can be read by measuring turbidity caused by a white precipitate of magnesium pyrophosphate without the need for gel electrophoresis. Moreover, its sensitivity is less affected than that of PCR by leaf components (Kaneko et al. 2007). For these reasons, the LAMP assay has been used to detect a range of plant pathogens (Harper et al. 2010; Kubota et al. 2008; Okuda et al. 2005; Oya et al. 2008).
In this study, we developed a LAMP assay for the detection and diagnosis of Stewart’s wilt from plant material in the field.
Materials and methods
Bacterial strains
We tested 26 strains of Pantoea spp., 17 strains of other genera (Table 1), and 57 unidentified strains isolated from the surface of maize leaves throughout Japan, the colonies of which all resemble Pantoea spp. on Luria–Bertani broth agar, Miller (Nakalai Tesque, Kyoto, Japan). All strains were used to test the specificity of the LAMP reaction. A pure culture of Pnss ICMP 5929 was used for optimizing the temperature for the LAMP reaction and for sensitivity and inoculation experiments. Sequence analysis of 5 strains of Pnss (ATCC 8199, ICMP 270, 722, 5929, 5930) was used to design LAMP primer sets. All strains were cultured in Luria–Bertani broth agar, Miller.
Sequencing analysis and primer design for LAMP
To obtain DNA sequences for designing LAMP primers, we performed conventional PCR. We used primer pair CPSL1/CPSR2c (Coplin et al. 2002) to amplify the cpsD region and PST3581/PST3909c (Wensing et al. 2010) to amplify the pstS–glmS region. To prepare template solutions, we suspended the bacteria in 1 mM 2-[4-(2-hydroxyethyl)-1-piperazinyl] ethanesulfonic acid (HEPES) solution (Nakalai Tesque). The bacterial cell suspensions (107 cfu/mL) were incubated at 98 °C for 10 min and then chilled on ice. The PCR using 5 µL of template solution was performed in a total volume of 20 µL containing 1 × Ex Taq buffer (20 mM Mg2+; Takara Bio, Ohtsu, Shiga, Japan), 200 µM dNTP mixture, 0.5 µM each primer, and 0.5 U of TaKaRa Ex Taq DNA polymerase (Takara Bio). PCR conditions were as described previously (Coplin et al. 2002; Wensing et al. 2010).
Loop-mediated isothermal amplification primers were designed from regions of cpsD and pstS–glmS highly conserved among the 5 Pnss isolates. The aforementioned PCR products were sequenced directly with a BigDye Terminator Kit v. 3.1 on an Applied Biosystems 3130 Genetic Analyser following the manufacturer’s instructions, and analysed with Sequencing Analysis v. 3.1 software (all from Applied Biosystems, Foster City, CA, USA). LAMP primers were designed from those sequences in PrimerExplore v. 4 software (Fujitsu Systems East, Tokyo, Japan). Two primer sets (PnsCps1 and PnsPst1) were designed (Table 2).
Conditions for LAMP reaction
Template solution was prepared as described for the conventional PCR. LAMP reactions using 5 µL of template solution were carried out with a Loopamp DNA amplification kit (Eiken Chemicals Co., Tokyo, Japan) in a 25-µL reaction mixture with a final concentration of 20 mM Tris·HCl (pH 8.8), 10 mM KCl, 8 mM MgSO4, 10 mM (NH4)2SO4, 0.1 % (v/v) Tween 20, 0.8 M betaine, 1.4 mM dNTPs, 8 U Bst DNA polymerase, 0.2 µM each outer primer (F3 and B3), and 1.6 µM each inner primer (FIP and BIP). To accelerate the reactions, 1.2 µM each of loop primers (loop-F, loop-B) were added to the reaction mixture (Nagamine et al. 2002).
For the surveillance of Stewart’s wilt at the disease-free area, Pnss-positive samples must be detected with certainty. For this purpose, it is desirable to use multiple primer sets that detect different genome sequences at the same time. Because the use of two primer sets at the same time in one incubator simplifies handling, the threshold time (T t, in min) of each primer sets was assessed at 60°, 63° and 65 °C to determine the optimal reaction temperature for both primer sets. The threshold was set for the time when the turbidity reached 0.1 (Okuda et al. 2008). The reaction mixture was incubated for up to 1 h in a Loopamp Real-Time Turbidimeter (LA-200, Teramecs, Kyoto, Japan).
Determination of sensitivity, specificity and tolerance to sweet corn leaf contents of LAMP
The sensitivity of LAMP and PCR was examined by using suspensions of Pnss of 106, 105, 104, and 103 cfu/mL in 1 mM HEPES solution. The sensitivity of LAMP was compared with that of PCR by using the pstS–glmS region-specific primer pair PST3899/PST4987c (Wensing et al. 2010). The conditions for LAMP were as described. The PCR reaction mixture was the same as described but with the addition of dimethyl sulfoxide to a final concentration of 5 % (v/v) and the following conditions: 94 °C for 2 min; 35 cycles at 94 °C for 20 s, 62 °C for 20 s, and 72 °C for 30 s; 72 °C for 2 min. These assays were performed independently 6 times. The specificity of LAMP was examined by using suspensions of all strains. These assays were performed independently 2 times.
To investigate the tolerance of LAMP to sweet corn leaf contents, the inhibitory effects of sweet corn leaf contents were compared using LAMP and PCR at 4 bacterial concentrations. To create a Pnss-free plant debris solution, a 50-mg section of a healthy leaf of sweet corn (cv. Honey Bantam) was sliced into pieces ≤1 mm thick, and the pieces were then soaked in 500 µL of 1 mM HEPES solution for 10 min; the supernatant was then used as the debris solution. Bacterial suspensions were diluted with the debris solution to 106, 105, 104, and 103 cfu/mL and used for LAMP and PCR assays as described above.
The usefulness of LAMP to detect Pnss in infected sweet corn leaves was tested. Sweet corn (cv. Honey Bantam) seedlings were inoculated at about the V4 stage by infusion of 10 µL of a suspension (108 cfu/mL) of Pnss into the base of the lowest leaf by pricking with a bundle of needles. After 2 weeks, the samples were prepared as described in the previous paragraph; we tested 40 samples that had water-soaked lesions on young, expanding leaves. Uninoculated leaves were used as negative controls. The extracts were incubated at 98 °C for 10 min and chilled on ice, then 5 µL was used as a template solution for LAMP.
Results and discussion
The five strains of Pnss showed 100 % nucleotide sequence identity in the amplified regions (accession numbers AB894425–AB894434). The PnsPst 1 primer set did not include loop-B primer, due to lack of suitable region for annealing.
To optimize the LAMP assay, we investigated the optimal temperature for the LAMP reaction. The mean T t values with the PnsCps1 primer set were increased in the order of 63 °C (19.6 min) < 65 °C (20.5 min) < 60 °C (21.7 min). Those with the PnsPst1 primer set were increased in the order of 60 °C (20.8 min) < 63 °C (21.6 min) < 65 °C (25.5 min). Analysis of variance followed by Tukey’s honestly significant difference test of pairwise comparisons showed that for the PnsCps1 primer set, there was a significant difference only between 63° and 60 °C, but for the PnsPst1 primer set, there were significant differences between all three temperatures (Fig. 1a, b). Although the difference in threshold time for the PnsPst1 primer set between 60° and 63 °C was statistically significant, the difference was only 1 min and for practical use is almost the same (Fig. 1b). Therefore, we decided that 63 °C is the optimal for the reaction temperature for practical use of the LAMP detection using both primers at the same time in one incubator. Further experiments were conducted using this temperature.

Threshold time (T t) values (time until turbidity = 0.1) (min) at 3 LAMP reaction temperatures (60°, 63°, 65 °C). a LAMP reaction with PsCps1 primer set. b Reaction with PsPst1 primer set. The long middle line is the group mean (n = 3); top and bottom lines represent the standard deviation
To confirm the effectiveness of the loop primers, we compared the reaction times with and without the loop primers. The addition of loop primers significantly shortened the mean T t values from 38.5 to 17.6 min (PnsCps1) and from 33.5 to 21.2 min (PnsPst1) (paired Student’s t test, P < 0.05; Fig. 2a, b).

Threshold time (T t) values (time until turbidity = 0.1) (min) with and without loop primers for LAMP reaction. a LAMP reaction with the PsCps1 primer set. b Reaction with the PsPst1 primer set. The long middle line shows the group mean (n = 3); top and bottom lines represent the standard deviation
The methods for the diagnosis should satisfy the requirements of International Standards for Phytosanitary Measures No. 27 for diagnostic protocols (IPPC 2006). Methods should be selected on the basis of their sensitivity, specificity and reproducibility. In addition, simple and quick methods are better for on-site inspection.
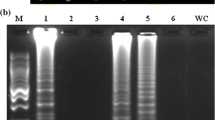
We also confirmed the LAMP sensitivity. The LAMP assays with both primer sets could detect all bacterial suspensions of 104 cfu/mL, with mean T t = 26.5 min (PsCps1) and 32.5 min (PsPst1), and maximum values of 28.7 min (PsCps1) and 33.3 min (PsPst1). On the other hand, DNA amplification from suspensions of 103 cfu/mL was not reliable (Table 3). Therefore, the detection threshold of the template bacterial suspension in this method was 104 cfu/mL. Conventional PCR could also detect as few as 104 cfu/mL. Thus, LAMP is at least as sensitive as conventional PCR. In the presence of leaf contents, LAMP could detect Pnss down to 104 cfu/ml, but conventional PCR failed to detect Pnss at any concentration (Table 4). This difference suggests that LAMP is better suited than conventional PCR for field surveillance of Stewart’s wilt. The tolerance of LAMP to the sweet corn leaf components obviates the need for pre-treatment of the template, reducing the time of the entire process to only 2 h.
The LAMP assay was specific to all Pnss strains, but it was negative for other Pantoea spp. strains and other genera associated with maize diseases (Table 1). In addition, no cross reaction was observed with 57 strains isolated from the surface of maize leaves in Japan. Another subspecies, P stewartii subsp. indologenes (Pnsi) (Mergaert et al. 1993), is commonly isolated from maize (Gehring et al. 2014), but it is a pathogen of millet and not virulent on maize (Mergaert et al. 1993). It is difficult to differentiate these subspecies by molecular methods. Unfortunately, we could not import strains of Pnsi, so we could not examine the specificity of the test for Pnss. But because Pnsi does not cause disease on maize, the LAMP assay is useful for identifying Pnss from leaves with disease symptoms. The identification of pure culture isolates or detection from asymptomatic leaves will still require additional tests such as virulence assays on seedlings, indole assays or subspecies-specific PCR (Gehring et al. 2014).
All 40 samples of tissue suspensions from fresh water-soaked lesions on young sweet corn seedling leaves on inoculated plants reacted positively with the two primer sets, but the samples from uninoculated leaves were negative. These results verify the reproducibility of the LAMP assay. They suggest that LAMP is suitable for on-site diagnosis and for high-throughput screening of Stewart’s wilt.
References
Coplin DL, Majerczak DR (1990) Extracellular polysaccharide genes in Erwinia stewartii: directed mutagenesis and complementation analysis. Mol Plant-Microbe Interact 3:286–292
Coplin DL, Majerczak DR, Zhang Y, Kim WS, Jock S, Geider K (2002) Identification of Pantoea stewartii subsp. stewartii by PCR and strain differentiation by PFGE. Plant Dis 86:304–311
Gehring I, Wensing A, Gernold M, Wiedemann W, Coplin DL, Geider K (2014) Molecular differentiation of Pantoea stewartii subsp. indologenes from subspecies stewartii and identification of new isolates from maize seeds. J Appl Microbiol 116:1553–1562
Harper SJ, Ward LI, Clover GRG (2010) Development of LAMP and real-time PCR methods for the rapid detection of Xylella fastidiosa for quarantine and field applications. Phytopathology 100:1282–1288
IPPC (International Plant Protection Convention) (2002) International Standards for Phytosanitary Measures: Diagnostic protocols for regulated pests. Pub No 27. Food and Agriculture Organization of the United Nations, Rome. http://www.ippc.int/sites/default/files/documents/20140428/ispm_27_2006_en_2014-04-28_201404281447–141.32%20KB.pdf. Cited 5 Nov 2014
Kaneko H, Kawana T, Fukushima E, Suzutani T (2007) Tolerance of loop-mediated isothermal amplification to a culture medium and biological substances. J Biochem Biophys Methods 70:499–501
Kubota R, Vine BG, Alvarez AM, Jenkins DM (2008) Detection of Ralstonia solanacearum by loop-mediated isothermal amplification. Phytopathology 98:1045–1051
Lamka GL, Hill JH, McGee DC, Braun EJ (1991) Development of an immunosorbent assay for seedborne Erwinia stewartii in corn seeds. Phytopathology 81:839–846
MAFF Plant Protection Station (2014) Plants and plant products subject to phytosanitary inspection at growing site. Internet Resource: http://www.pps.go.jp/english/faq/import/seeds.html. Cited 19 Sep 2014
Mergaert J, Verdonck L, Kersters K (1993) Transfer of Erwinia ananas (synonym, Erwinia uredovora) and Erwinia stewartii to the genus Pantoea emend. as Pantoea ananas (Serrano 1928) comb. nov. and Pantoea stewartii (Smith 1898) comb, nov., respectively, and description of Pantoea stewartii subsp. indologenes subsp. nov. Int J Syst Evol Microbiol 43:162–173
Nagamine K, Hase T, Notomi T (2002) Accelerated reaction by loop-mediated isothermal amplification using loop primers. Mol Cell Probes 16:223–229
Notomi T, Okayama H, Masubuchi H, Yonekawa T, Watanabe K, Amino N, Hase T (2000) Loop-mediated isothermal amplification of DNA. Nucleic Acids Res 28:E63
Okuda M, Matsumoto M, Tanaka Y, Subandiyah S, Iwanami T (2005) Characterization of the tufB-secE-nusG-rplKAJL-rpoB gene cluster of the citrus greening organism and detection by loop-mediated isothermal amplification. Plant Dis 89:705–711
Okuda M, Kawano S, Murayama Y, Iwanami T (2008) Conditions for loop-mediated isothermal amplification (LAMP) and a nonmacerating DNA extraction method to assay for huanglongbing (citrus greening) disease (in Japanese with English summary). Jpn J Phytopathol 74:316–320
Oya H, Nakagawa H, Saito N, Uematsu H, Ohara T (2008) Detection of Acidovorax avenae subsp. citrulli from seed using LAMP method (in Japanese with English summary). Jpn J Phytopathol 74:304–310
Pataky J, Ikin R (2003) Pest risk analysis: the risk of introducing Erwinia stewartii in maize seed. International Seed Federation, Geneva. http://www.worldseed.org/cms/medias/file/TradeIssues/PhytosanitaryMatters/PestRiskAnalysis/Erwinia_stewartii.pdf. Cited 5 Nov 2014
Tambong JT, Mwange KN, Bergeron M, Ding T, Mandy F, Reid LM, Zhu X (2008) Rapid detection and identification of the bacterium Pantoea stewartii in maize by TaqMan real-time PCR assay targeting the cpsD gene. J Appl Microbiol 104:1525–1537
Wensing A, Zimmermann S, Geider K (2010) Identification of the corn pathogen Pantoea stewartii by mass spectrometry of whole-cell extracts and its detection with novel PCR primers. Appl Environ Microbiol 76:6248–6256
Xu R, Chen Q, Djama ZR, Tambong JT (2010) Miniprimer PCR assay targeting multiple genes: a new rapid and reliable tool for genotyping Pantoea stewartii subsp. stewartii. Lett Appl Microbiol 50:216–222
Acknowledgments
We thank the National Institute of Agrobiological Sciences for providing the bacterial strains.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Uematsu, H., Inoue, Y. & Ohto, Y. Detection of Pantoea stewartii from sweet corn leaves by loop-mediated isothermal amplification (LAMP). J Gen Plant Pathol 81, 173–179 (2015). https://doi.org/10.1007/s10327-015-0580-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10327-015-0580-4