
Abstract
Marine sediments from Newfoundland, Canada were explored for biotechnologically promising Actinobacteria using culture-independent and culture-dependent approaches. Culture-independent pyrosequencing analyses uncovered significant actinobacterial diversity (H′—2.45 to 3.76), although the taxonomic diversity of biotechnologically important actinomycetes could not be fully elucidated due to limited sampling depth. Assessment of culturable actinomycete diversity resulted in the isolation of 360 actinomycetes representing 59 operational taxonomic units, the majority of which (94 %) were Streptomyces. The biotechnological potential of actinomycetes from NL sediments was assessed by bioactivity and metabolomics-based screening of 32 representative isolates. Bioactivity was exhibited by 41 % of isolates, while 11 % exhibited unique chemical signatures in metabolomics screening. Chemical analysis of two isolates resulted in the isolation of the cytotoxic metabolite 1-isopentadecanoyl-3β-d-glucopyranosyl-X-glycerol from Actinoalloteichus sp. 2L868 and sungsanpin from Streptomyces sp. 8LB7. These results demonstrate the potential for the discovery of novel bioactive metabolites from actinomycetes isolated from Atlantic Canadian marine sediments.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The combination of microbial evolution and genetic transfer between species has resulted in the emergence of new pathogens and an increase in antibiotic-resistant strains, resulting in an urgent need for the discovery of new therapeutic agents [18]. Furthermore, the requirement for efficacious treatments against cancer and infectious disease continues to drive the discovery of novel bioactive metabolites [5, 41]. Bacteria belonging to the order Actinomycetales account for the production of over 45 % of all bioactive microbial metabolites. Within this order, the genus Streptomyces is unsurpassed for the production of biotechnologically relevant bioactive secondary metabolites, producing over 7,600 of the greater than 10,000 bioactive compounds discovered from actinomycetes [5]. Over the last 50 years, bioprospecting from the Oceans has uncovered numerous novel metabolites from marine actinomycetes. These discoveries include salinosporamide A, a potent proteasome inhibitor produced by Salinispora, abyssomycin C, a polycyclic antibiotic from a Verrucosispora strain and lajollamycin, a potent lactam antibiotic from a Streptomyces strain [22, 47, 64].
The marine environment comprises a plethora of varied ecosystems encompassing extensive metabolic and genetic diversity [7, 44, 74]. Parameters including tidal variation, nutrient availability, temperature and salinity greatly influence the diversity of organisms found in a specific environment [8, 28]. Isolation of novel bacteria from underexplored marine ecosystems can greatly enhance the potential to discover novel bioactive secondary metabolites [8, 10, 17, 43]. The concept of biodiversity translating to chemical diversity is widely accepted; therefore, isolation of novel bacteria from as yet unexplored locations is a strategy with significant potential for the discovery of novel bioactive chemicals [5, 44, 77]. The biogeographical hypothesis “everything is everywhere, but the environment selects” and the influence of environmental conditions on microbial distribution has been considered [16, 36]. There is increasing evidence to link the physiology and ecology of microorganisms with biogeographical distribution of taxa [24]. Several reports have recently investigated the geographic effects on species distribution of marine planktonic diatoms [11], spore-forming thermophillic bacteria in Arctic marine sediment [31] and Arctic microbial communities [13] with some studies reporting an effect on microbial diversity that has been observed with spatial distribution [48, 63, 83]. Freel et al. suggested that secondary metabolism influences ecological diversification within the genus Salinispora, where distinct species distribution is linked to specific geographic location and species-specific secondary metabolite profiles have been observed [24, 38]. Actinomycetales, have been successfully cultured from the marine environment from varied habitats including the Mariana Trench [60], tropical sediments [29, 33, 34, 45, 50], sponges [53], North Sea sediments [82] and sediment from a Norwegian fjord [8]. Despite improved culture methods, it is estimated that >99 % of the total bacterial diversity within an environmental sample cannot be cultured using standard culture methods [37, 46]. The diversity of culturable actinomycetes from tropical environments has been studied to a greater extent than that of temperate marine habitats and actinomycete diversity in the sediments of Atlantic Canada remains largely unexplored. Consequently, the isolation of actinomycetes from the coast of Newfoundland (NL), Canada provides an opportunity to isolate biologically and chemically diverse actinomycetes.
The extent of bacterial diversity within the marine environment has only recently been revealed using culture-independent techniques. Significant advances in culture-independent molecular techniques allow for an in-depth assessment of bacterial diversity. Specifically, 454-massively parallel pyrosequencing of metagenomic 16S rRNA amplicons allows large sequence libraries to be generated, detailing the bacterial community composition of targeted environments. Next-generation sequencing technology has uncovered exceptional levels of microbial diversity in habitats as varied as forest and grassland soils [54], the deep sea [74], marine invertebrates [76] and Arctic soils [13]. The knowledge of bacterial taxonomic diversity present in specific environmental sample can be utilized to engineer taxa-selective isolation approaches and focus on underexplored environments that have the potential to yield both large numbers and diverse assemblages of biotechnologically desirable bacteria, such as actinomycetes. In this study, the complementary nature of culture-independent and culture-dependent approachs was used to fully understand microbial diversity and explore the biotechnological potential of actinomycetes. Pyrosequencing was used to determine both actinobacterial and total bacterial diversity within sediments collected from eight locations around Bonne Bay, NL. A complimentary culture-based approach was used to evaluate the cultivable diversity of actinomycetes and to establish a library of filamentous actinomycetes from each of the eight sediment samples. Furthermore, the metabolic potential of the library was assessed through a combination of antimicrobial and anticancer biological screening as well as metabolomics-based chemical screening.
Materials and methods
Sample collection and processing
Eight sediment samples were collected in August 2009 from intertidal and subtidal zones in and around Bonne Bay, NL, Canada. Four subtidal sediment samples were collected by self-contained underwater breathing apparatus from different areas within Bonne Bay and are named 1L (49.489090, −57.911682, depth 12.5 m), 2L (49.537028, −57.917861, depth 2.5 m), 3L (49.515300, −57.850417, depth 9 m) and 6L (49.466317, −57.728450, depth 10.5 m). Four intertidal sediments were collected by snorkelling: 4L (49.923583, −57.777167, depth <0.5 m), 5L (49.858600, −57.825333, depth 2.5 m), 7L (49.606500, −57.951000, depth 1.8 m) and 8L (49.559967, −57.832317, depth <0.5 m). Samples were collected aseptically using a sterile scoopula and 50 mL conical tube. Samples were transported to the laboratory on ice (≤4 h), after which a portion (~2 g) of each sample was immediately frozen at −80 °C for future DNA extraction and the remainder was processed for actinomycete culturable diversity studies the same day.
Isolation of sediment metagenomic DNA
Metagenomic DNA was isolated from sediment samples (~0.5 g) using the Fast DNA Spin Kit for Soils (MP Biomedicals, Solon, OH, USA) as described previously [21] and stored at −20 °C. Concentration and integrity of isolated DNA was determined by UV spectroscopy and agarose gel electrophoresis (1 % agarose, 1X Tris–acetate-EDTA buffer stained with ethidium bromide) [39].
16S rRNA amplicon pyrosequencing and sequence analysis
Bacterial diversity was assessed by pyrosequencing of 16S rRNA amplicons generated from sediment metagenomic DNA. Bacterial tag-encoded FLX amplicon pyrosequencing (bTEFAP) of metagenomic 16S rRNA amplicons was performed by Research and Testing Laboratory (Lubbock, TX, USA) as previously described [1] based upon RTL protocols (http://www.researchandtesting.com). bTEFAP utilizes Titanium reagents and procedures with a one-step PCR reaction containing a mixture of both HotStart and HotStart high fidelity Taq polymerases and amplicons originating from the 27F region numbered in relation to E. coli rRNA [20].
Raw pyrosequencing data was analyzed using mothur v.1.32 according to published recommendations [67, 68] and as described previously [21] with the exception that sequences were removed from the analysis if they were shorter than 250 bp. Operational taxonomic units (OTUs) were defined at a 3 % distance as previously described [67]. Observed and estimated richness (Chao1), sampling coverage (C), Shannon evenness (E) and diversity (H′) indices and rarefaction curves were calculated using mothur [67]. To calculate alpha diversity statistics for the actinobacterial portion of the sediment sequence libraries, Actinobacteria (phylum)-affiliated sequences were first obtained from the final quality filtered sequence library using the get.lineage command in mothur. The actinobacterial sequences were then clustered into OTUs (D = 0.03) and the diversity statistics calculated as described above. Sequences were archived through the NCBI sequence read archive (accession numbers: SS1460687, SS1460688, SS1460709–SS1460714).
Actinomycete culturable diversity
A combination of sediment pre-treatments, plating techniques and selective isolation media were employed to selectively isolate mycelia-forming actinomycetes. The dry/stamp (DS) method involved drying sediment overnight in a laminar flow hood. Dried sediments were subsequently stamped onto the surface of agar plates using a sterile foam plug (2 cm diameter) eight times to create a serial dilution effect [34]. The dilute/heat (DH) method involved diluting 1 mL of wet sediment with 10 mL sterile seawater and then heating the diluted sediment to 55 °C for 6 min. A 200 μL aliquot was used to inoculate each agar plate [34]. The direct plating (DP) method consisted of spreading 1 g of sediment directly on to each agar plate. For each pre-treatment and plating combination, five isolation media were used: Starch Casein Nitrate agar (SC), Raffinose-Histidine agar (RH) [45], Chitin Low Nutrient agar (CH) [51], Phosphate-Nitrate agar (PN) [46] and Actinomycete Isolation agar (AC) [3]. All media were prepared with filtered natural seawater (collect from the Southern coast of Prince Edward Island, Canada) and sterilized by autoclaving. To reduce the growth of fungi and non-actinomycete bacteria, media were supplemented with nystatin (AC—10 µg mL−1; RH, CH, SC, PN—50 µg mL−1); cycloheximide (AC—50 µg mL−1; RH, CH—100 µg mL−1) and nalidixic acid (SC, PN—10 µg mL−1). Four replicates of each pre-treatment (DH, DS, DP) and media (RH, SC, AC, CH, PN) combination were plated resulting in 60 isolation plates per sediment and 480 plates in total. Plates were monitored for the emergence of colonies exhibiting morphology typical of filamentous actinomycetes (leathery substrate mycelia and formation of aerial mycelia and spores) for 3 months. Colonies exhibiting the targeted morphology were chosen for purification and initially subcultured onto the same media from which they were isolated, but prepared with 18 g L−1 Instant Ocean® (IO; Spectrum Brands, Middleton, WI, USA) and Milli-Q water to replace seawater. Isolates were recursively subcultured onto International Streptomyces Project Medium 3 (ISP-3) [3] supplemented with 18 g L−1 IO until pure. Isolates were dereplicated within sample location based on morphological characteristics (color and texture of both aerial and substrate mycelia, production of soluble pigments and presence/absence/color of spores). Cultures were archived as spore suspensions or vegetative mycelial stocks in 20–25 % glycerol (v/v) and stored at −80 °C.
Identification of bacterial isolates
Axenic isolates were identified as described previously [21]. Briefly, 16S rRNA genes were amplified using EconoTaq® PLUS GREEN 2X Master Mix (25 µL) (Lucigen, Middleton, WI, USA) and the primers pA (5′-AGAGTTTGATCCTGGCTCAG) and pH (5′-AAGGAGGTGATCCAGCC) [81]. Partial sequencing of 16S rRNA amplicons employed the 16S530R primer (5′-GTATTACCGCGGCTGCTGG) [29]. In some cases full-length 16S rRNA gene sequencing was performed using the additional primers 16S936R (5′-GGGGTTATGCCTGAGCAGTTTG), 16S1527R (5′-AAGGAGGTGATCCAGCC), 16S514F (5′-GTGCCAGCASCCGCGG) and 16S1114F (5′-GCAACGAGCGCAACCC) [29].
Phylogenetic analysis of cultured actinomycetes
Sequences (~530 bp) were analyzed, edited and grouped using the Contig Express application within the Vector NTI software package (v.10.3, Invitrogen, Carlsbad, CA, USA). Isolates were dereplicated based on 16S rDNA gene sequence identity. Isolates sharing ≥99 % sequence identity were grouped into OTUs and a representative sequence from each group was used for subsequent analysis. Isolates termed “individuals” (IND) were OTUs comprised of a single isolate. Sequences of OTU representatives were compared to sequences within the NCBI database (http://blast.ncbi.nlm.nih.gov/Blast.cgi) using the Basic Local Alignment Search Tool (BLASTN) [2]. Phylogenetic analysis of partial 16S rRNA gene sequences was conducted using Bionumerics (Version 6.1, Applied Maths) as described previously [21]. The partial 16S rRNA gene sequences of 59 OTUs were deposited in the GenBank nucleotide sequence database under the accession numbers KM102181–KM102246 and KM111605.
Small-scale fermentation and extraction
Inocula for fermentations were prepared using a two-stage seed culture approach. Cultures from agar plates were used to inoculate 7 mL of ISP2 broth supplemented with 18 g L−1 IO in culture tubes (150 × 25 mm) containing 3–5 glass beads. First stage seed cultures were grown at 30 °C with shaking at 200 rpm for 72 h. A portion (1 mL) of this culture was used to inoculate a culture tube containing fresh medium (7 mL). This second-stage seed culture was incubated under identical conditions for 24 h. Fermentations (50 mL medium in a 250 mL Erlenmeyer flask) were inoculated with 3 % v/v of the second-stage seed culture and incubated under identical conditions for 7 days.
The formulations of the ten fermentation media used in this study are summarized in Table S1. All media contained 18 g L−1 IO except for ISP2 medium, which was used as fermentation medium for metabolomics analysis. ISP2 was prepared without IO because all strains were able to grow in the absence of salt, and its omission enhanced subsequent chemical analysis of fermentation extracts by reducing levels of coextracted salt. Activated Diaion HP20 resin (5 % w/v) (Itochu Chemicals America, White Plains, NY) was added to all fermentation media other than ISP2 prior to sterilization. All media were sterilized by autoclaving at 121 °C for 15 min. Not all strains were fermented in all media. Four strains (marked with * in Table 2) were fermented in all media except ISP2. One strain was fermented in all 10 media (marked with a ^ in Table 2). Four strains were fermented in five media (BFM1-4 and ISP2; marked with a ╪ in Table 2). An additional 23 strains were only fermented in ISP2 (unmarked in Table 2).
After 7 days of growth Amberlite® XAD7HP resin (5 % w/v) (Sigma, Oakville, ON, Canada) was added to fermentations (except those conducted in ISP2) and agitated for 1 h at room temperature. The cells and resin were collected via centrifugation (4,000×g, 10 min) and washed twice with MilliQ water (40 mL). Pellets were frozen (−80 °C overnight), lyophilised and then thrice extracted with 10 mL of methanol (MeOH) by rapid agitation (200 rpm) for 1 h. Extracts were dried in vacuo and fractionated using C18 HyperSep™ SPE (Solid Phase Extraction) SepPack columns (6 mL volume, 500 mg bed weight) (Thermo Scientific, Mississauga, ON, Canada) and aqueous MeOH step gradients. Three fractions (F1-3) were generated corresponding to 10, 50 and 100 % MeOH, respectively. Fractions and crude extracts were tested for bioactivity. Fermentations conducted in ISP2 for metabolomics analysis were extracted with 10 mL ethyl acetate (EtOAc) by rapid agitation (200 rpm) at room temperature for one hour. Extracts were dried in vacuo and defatted by partitioning between 8:2 acetonitrile (ACN):H2O and hexanes (10 mL each). Defatted extracts (ACN:H2O fraction) were tested for bioactivity.
Antimicrobial testing
Microbroth assays were conducted in 96-well plates according to the Clinical Laboratory Standards Institute testing standards [56]. Extracts were evaluated for antibiotic activity against methicillin-resistant Staphylococcus aureus ATCC 33591, vancomycin-resistant Enterococcus faecium EF379, Proteus vulgaris ATCC 12454 and Candida albicans ATCC 14035. Fractionated extracts were dissolved in sterile 20 % dimethyl sulfoxide (DMSO) and tested at a final concentration of 50 µg mL−1. Crude extracts were tested at 20 and 100 µg mL−1. Extracts were considered active if they exhibited >70 % growth inhibition.
Cytotoxicity testing against primary breast cancer cell line HMT3909S8
Crude fermentation extracts of nine strains (Table 2) were tested for growth inhibition of the primary breast cancer cell line HMT3909S8 at a concentration of 20 μg mL−1. Testing was carried out at AvantiCell Science Limited (Ayr, Scotland). A negative control (cells treated with 1 % DMSO solvent control), 100 % lysis control (cells treated with 0.18 % Triton X-100), positive controls (actinomycin D and staurosporine, 10 μM each) and a blank (cell-free wells with assay reagent mix added to adjust for non-specific background) were tested in triplicate in 96-well plates. Test samples were added to culture wells during the final 24 h of the 96 h incubation. Cytotoxicity was measured as the extracellular appearance of cytosolic dehydrogenase enzymes using a coupled fluorimetric enzymatic reaction. Fluorescence intensity was measured using a fluorescent plate reader with excitation and emission wavelengths set to 530 and 590 nm, respectively. Cytotoxicity was calculated by expressing the fluorescence intensity of each test well as a percentage of the mean florescence intensity of fully lysed cells in 100 % lysis control wells. Specific lytic activity of samples was calculated by dividing the percent lysis by the sample test concentration. Samples exhibiting specific lytic activity >1 were considered active.
Cytotoxicity testing against established cell lines
Human BJ fibroblast cells (ATCC CRL-2522) were grown and maintained in Eagle’s minimal essential medium supplemented with fetal bovine serum (10 %), penicillin (100 µU) and streptomycin (0.1 mg mL−1). Human breast adenocarcinoma cells (ATCC HTB-26) were grown and maintained in Dulbecco’s Modified Eagle’s Medium/Nutrient Mixture F-12 Ham supplemented with fetal bovine serum (10 %), penicillin (100 µU) and streptomycin (0.1 mg mL−1). The cells were incubated (37 °C, humidified atmosphere, 5 % CO2). Culture media were refreshed every 2–3 days and cells were not allowed to exceed 80 % confluence. At 80 % confluence, the cells were counted, diluted and plated into 96-well treated cell culture plates. The BJ fibroblast and HTB-26 cells were plated in 90 µL of respective growth medium (10,000 and 5,000 cells/well, respectively). All media used was as previously described without the addition of antibiotics. The plates were incubated as before to allow cells to adhere to the plates for 24 h before treatment. DMSO was used as the vehicle at a final concentration of 1 %. The fibroblasts and the HTB-26 cells were incubated as previously described for 24 and 72 h, respectively. All samples were tested in triplicate. Each plate contained four un-inoculated media blanks (media + 1 % DMSO), four untreated growth controls (media + 1 % DMSO + cells), and one column containing serially diluted zinc pyrithione or doxorubicin as positive controls. AlamarBlue (Life Technologies, Carlsbad, CA, USA) was added to each well 24 h after treatment (10 % v/v). Fluorescence (560/12 excitation, 590 nm emission) was monitored using a Varioskan Flash Multimode plate reader (Thermo Scientific, Mississauga, ON, Canada) both at time zero and 4 h after the addition of alamarBlue. After subtraction of fluorescence at time zero from 4 h readings the percentage of cell viability relative to vehicle control wells was calculated.
High-resolution mass spectrometry analysis
Extracts and fractions were analyzed by ultra high performance liquid chromatography-high-resolution mass spectrometry (UPLC-HRMS) using an Orbitrap Exactive (Thermo Fisher Scientific, Mississauga, ON, Canada) UPLC system with a Kinetex 1.7 μm C18 100 Å 50 × 2.1 mm column (Phenomenex, Torrance, CA, USA). A linear gradient from 95 % H2O/0.1 % formic acid (FA) (solvent A) and 5 % ACN/0.1 % FA (solvent B) to 100 % solvent B over 5 min followed by a hold of 100 % solvent B for 3 min was used with a flow rate of 400 μL/min. Eluate was detected by +ESI-HRMS monitoring m/z 200–2,000 amu, evaporative light scattering detector (ELSD; Sedex; Sedere, Alfortville, France) and PDA (200–600 nm).
Identification of cytotoxic metabolites produced by Actinoalloteichus sp. 2L868
Actinoalloteichus sp. 2L868 was fermented in 27 fernbach flasks containing 600 mL of BFM2 medium (16.2 L total). A two-stage seed protocol was used as described previously and fermentations were inoculated with 3 % v/v of the second-stage seed culture. Fermentations were incubated at 30 °C and 200 rpm for 12 days. Cells and resins were collected by centrifugation and extracted as previously described using ethanol. The crude extract was partitioned between H2O and EtOAc. The EtOAc extracts were concentrated by rotary evaporation to obtain a dark brown oily extract (301.5 mg), which was subsequently partitioned between ACN:H2O (8:2) and hexane. Resulting hexane (115.5 mg) and ACN (174.7 mg) fractions were evaluated for antimicrobial and cytotoxic activity to confirm that the scaled-up fermentation retained biological activity. The ACN fraction was separated by flash chromatography (Combiflash Rf, Teledyne Isco, Lincoln, NE, USA; C18 column, 48 g) using a 5 min isocratic mixture of MeOH:H2O 1:9, followed by a 20 min continuous gradient until 100 % MeOH, then 10 min 100 % isocratic MeOH (40.0 mL/min). Twenty-two fractions were collected by UV trace (254 nm). Five non-polar fractions showed considerable antimicrobial and cytotoxic activities. Each fraction was separated using a Waters HPLC system (Synergi Fusion RP 4 µm, 10 × 250 mm column; Phenomenex, Torrance, CA, USA) using a linear gradient from 50 % MeOH/0.1 % FA (solvent A) and 50 % H2O/0.1 % FA (solvent B) to 100 % solvent A over 30 min, followed by 20 min of 100 % solvent A (flow rate 3.0 mL/min). HPLC fractions were analyzed by UPLC-HRMS as described above. Compounds were identified using an NMR spectrometer (Bruker Avance 600 MHz). Spectral data from 1D and 2D NMR experiments and UPLC-HRMS analysis were compared to literature data to confirm the identity of known compounds.
Metabolomic analysis of small-scale fermentations
Metabolomic analysis of fermentation extracts was conducted as previously reported [23]. Briefly, fermentation extracts were analyzed by UPLC-HRMS as described above. MZmine 2 [61] was used for peak picking of UPLC-HRMS profiles, set with an intensity threshold of 5E5 units, followed by deisotoping, bucketing alignment, standardization and artifact suppression. Cluster analysis and principal component statistical analyses were conducted using The Unscrambler (Camo Software, Woodbridge, NJ, USA). Compounds identified via metabolomics analysis were purified using a Waters HPLC system (Milford, MA, USA) with a semi preparative column (Gemini 5 μm C18 110 Å 250 × 10 mm; Phenomenex, Torrance, CA, USA) and eluate was monitored by ELSD and PDA (254 and 280 nm). Compounds were identified using an NMR spectrometer (Bruker Avance 600 MHz). Spectral data from 1D and 2D NMR experiments and UPLC-HRMS analysis were compared to literature data to confirm the identity of known compounds [65].
Purification of sungsanpin
Sungsanpin [78] was purified by reverse phase flash chromatography and RP-HPLC using LCMS to monitor for m/z 1,592.8184. Crude extracts were fractionated using a 13 g C18 column with a linear gradient from 9:1 H2O:MeOH to 100 % MeOH over 13 min followed by 100 % MeOH hold for 5 min. Sungsanpin eluted at 12.5 min. 3.6 mg of Sungsanpin was purified using a Waters HPLC system with a Gemini 110A C18 5 µm 250 × 10.00 mm column (Phenomenex, Torrance, CA, USA). Sungsanpin was eluted with H2O/0.1 % FA (solvent A) and MeOH/0.1 % FA (solvent B) with a flow rate of 3 mL/min and a linear gradient increasing from 50 % solvent A and 50 % solvent B to 20 % solvent A and 80 % solvent B over 17 min, followed by a linear increase to 100 % solvent B over 2 min, followed by 100 % solvent B for 12 min. Sungsanpin eluted at 14.9 min and was monitored by ELSD and UV at 220 and 280 nm. UPLC-HRMS was used to screen fractions for m/z 1,592.8184. MS/MS analysis was conducted by direct infusion using an LTQ Orbitrap Velos (Thermo Scientific, Mississauga ON, Canada) at a rate of 2 µL min−1 using +ESI followed by isolation of the [M + H] in the linear ion trap and fragmented by higher energy collisional dissociation with an energy of 29 eV.
Human and animal rights
No humans or animals were affected by the research presented here.
Results
Culture-independent analysis of bacterial diversity in NL marine sediments
Processing of raw pyrosequencing data using mothur yielded a total of 39,761 high quality partial 16S rDNA sequence reads. Sampling depth in each sample ranged from 2,681 to 6,858 sequences and the average sequence length was 251 bp (Table 1). Phylum level bacterial communities were dominated by Bacteroidetes (14.10–56.1 %), Proteobacteria (25.44–59.0 %) and unclassified Bacteria (12.5–33.4 %). Collectively, these three groups accounted for 85.6–95.8 % of sequences obtained from each sediment. Despite the relatively large sampling depth obtained via pyrosequencing in this study compared to traditional clone libraries, a limited number of actinobacterial sequences were detected. The number of actinobacterial sequences in each sediment varied from 21 to 354 sequences, comprising 0.3–5.9 % of each sequence library (Table 1). Within the class Actinobacteria the dominant orders were the Acidimicrobiales (4–51 %), Actinomycetales (9–91 %) and unclassified Actinobacteria (3–43 %) (Table S2).
To compare overall bacterial and actinobacterial diversity a variety of alpha diversity statistics were calculated. In terms of overall bacterial diversity, observed (S obs) and predicted species level richness (S est-, Chao1 estimate, [12] ) varied from 641 to 1,382 OTUs and 1,544 to 3,625 OTUs, respectively (Table 1). Overall bacterial diversity in the sediments was high with calculated Shannon diversity (H′) and equitability (E) index values ranging from 4.95 to 6.68 and 0.77–0.93, respectively (Table 1). Due to the limited number of actinobacterial sequences detected in the NL sediments the observed and predicted species richness (S obs 13–54 OTUs, S est 29–109 OTUs) was understandably low (Table 1). Actinobacterial diversity calculated using the Shannon diversity index ranged from 2.45 to 3.54, which although lower than the values obtained for the entire bacterial community, still indicates significant species level diversity (Table 1). To assess the adequacy of sampling depth Good’s coverage (C) was calculated. For overall bacterial communities the sampling depth achieved in this study was adequate to detect 64–84 % of the species level richness (Table 1). In comparison, the sampling depth of actinobacterial sequences was only sufficient to detect >64 % of OTUs in three of eight sediments (4L, 6L and 8L), with the coverage for the remaining five samples ranging from 37 to 54 % (Table 1). Rarefaction analysis also indicated that additional sampling would likely reveal additional actinobacterial diversity as rarefaction curves did not reach an asymptote (Fig. S1).
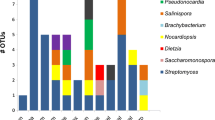
Due to the well-known ability of taxa belonging to the order Actinomycetales to produce bioactive natural products (NPs) [5], we further explored taxonomic affiliations of actinomycetes present in the NL sediments (Fig. 1; Table S3). Actinomycetes which could not be assigned to a family (using an 80 % confidence threshold) were the dominant actinomycetes, constituting 33.3–100 % of sequences in each sediment. Microbacteriaceae were present (0.3–12.3 %) in four sediment samples (1L, 4L, 6L, 8L). Mycobacteriaceae were present in two samples (1L, 55.6 % and 7L, 33.3 %) while Nocardioidaceae (4L, 26.3 %), Geodermatophilaceae (6L, 11.1 %), Kineosporiaceae (8L, 45.3 %) and Propionibacteriaceae (4L, 1.8 %) were only detected in a single sample.

Family level bacterial community composition within the order Actinomycetales of eight NL sediment samples. Numbers following sample name in brackets indicate the number of sequences from each sample classified as Actinomycetales
Culture-dependent actinomycete diversity
Over 1,500 isolates displaying typical morphological characteristics of filamentous actinomycetes were initially isolated and dereplicated to 360 morphologically unique isolates for partial 16S rRNA gene sequencing. The number of strains sequenced from each location were as follows: 1L-43, 2L-51 3L-51, 4L-45, 5L-23, 6L-55, 7L-47, 8L-45. The DS pre-treatment was the most successful for isolating filamentous actinomycetes (151), followed by DP (131) and DH (79). SC media yielded the highest number of actinomycete isolates (119), followed by RH (71), AC (70), PN (59) and CH (41). OTUs were defined using a ≥99 % sequence identity threshold as previous research has shown that in Actinobacteria 16S rDNA sequence identities <99 % correlate well with DNA–DNA reassociation values of <70 %, the value commonly used to discriminate between species [75]. Using this OTU definition 59 OTUs were identified and used in all subsequent analyses (Tables S4 and S5). Isolates were identified by BLASTN comparison to sequences contained in GenBank and the phylogenetic relationships between the OTUs and selected reference strains were analysed by construction of a NJ tree (Fig. 2).

Neighbor-joining tree showing the phylogenetic relationship of cultured actinomycetes obtained from NL sediments. The numbers at the nodes indicate the degree of bootstrap support (%) based on 1,000 iterations, bootstrap values >50 % are shown. The scale bar represents percent sequence similarity. Numbers in brackets represent the following: number of isolates in clade—number of OTUs in clade—number of OTUs represented by a single isolate (singleton). Collection site(s) (e.g. 1L–8L) represented in each clade are shown after the brackets
Actinomycete isolates formed five major clades representing the genera Promicromonospora (Fig. 2—Clade A; 1 isolate), Actinoalloteichus (Fig. 2—Clade B; 1 isolate), Nocardiopsis (Fig. 2—Clade C; 21 isolates comprising 4 OTUs) and Streptomyces (Fig. 2—Clade D and E). Clades containing different genera were supported by high bootstrap values (>99 %). The major Streptomyces clades (D and E) were divided into five subclades (D1, D2, E1, E2 and E3). Differentiation of Streptomyces clades D and E was well supported (93 %), while the subclades were supported by bootstrap values <70 %, suggesting that although all isolates were assigned to the Streptomyces genus, some subclades lacked robust assignment within the order of the phylogenetic tree. The Streptomyces subclade D1 included seven strains (two OTUs) from four sites, while 110 strains (12 OTUs total, six singletons) originating from all sites comprised subclade D2. Subclade E1 encompassed one OTU containing three isolates from 5L and 6L; E2 consisted of 25 isolates belonging to seven OTUs, three of which were singletons, originating from all but two sites. Subclade E3, the largest Streptomyces clade, contained 192 isolates (31 OTUs total, 20 singletons) originating from all sites. Due to the limited resolving power of partial 16S rDNA sequences, fine scale phylogenetic variation within the subclades could not be accurately determined as bootstrap support for branches within the subclades was low.
Bioactivity screening of selected actinomycetes
To explore the biotechnological potential of the actinomycetes purified from NL sediments, a selection of 32 isolates were fermented in a variety of media and the resulting extracts tested for biological activity (Table 2). Isolates were chosen from all clades in Fig. 2 (with the exception of clade A) and represented a variety of OTUs from within each clade. The majority of isolates tested were Streptomyces with the exception of a single Actinoalloteichus isolate (2L868) and two Nocardiopsis isolates (6LB3 and 2LD8).
Extracts from nine isolates belonging to clades B, D2, E2, and E3 in Fig. 2 were tested for cytotoxicity against the human primary breast cancer cell line HMT3909S8 (Table 2). The level of activity considered a hit in this assay was a specific lytic activity (SLA) >1. Significant lytic activity was exhibited by extracts of Actinoalloteichus sp. 2L868 (clade B) fermented in BFM1 (30.6 % lysis, SLA—1.53) and BFM2 (34.8 % lysis, SLA—1.74). Extracts of Streptomyces isolates 6LA4 and 3LE1 (both clade D2) fermented in BFM3 exhibited lytic activity of 24.0 % (SLA—1.2) and 23.2 % (SLA—1.2), respectively. The lytic activity of these extracts greatly exceeded that obtained for the positive controls actinomycin D (2.10 % lysis) and staurosporine (10.50 % lysis).
Fermentation extracts from all 32 isolates were tested for antimicrobial activity against the Gram-positive pathogens methicillin-resistant S. aureus (MRSA) and vancomycin-resistant E. faecalis (VRE), the Gram-negative pathogen P. vulgaris, and the fungus C. albicans (Table 2). Forty-one percent of isolates (13) exhibited antimicrobial activity (>70 % growth inhibition) against one or more pathogens. Six isolates (18.8 %) exhibited antimicrobial activity against MRSA and VRE, one isolate (3.1 %) was active against P. vulgaris, and ten isolates (31.3 %) were active against C. albicans. The antimicrobial activity was produced by isolates belonging to clades B, D2, E2 and E3 (representative of clade A were not tested). No bioactivity was exhibited by extracts of the two Nocardiopsis isolates (clade C) or the sole streptomycete representatives of clades D1 and E1. Of the 13 isolates exhibiting antimicrobial activity, approximately, half were active against multiple test organisms (2L868, 6LA4, 3LD1, 57A7, 2LD2 and 8LD1). Activity against multiple organisms could be due to the production of multiple bioactive metabolites or the production of a single metabolite with broad spectrum activity.
The activity against P. vulgaris by a single isolate (5LA7, 71 % inhibition at 50 μg mL−1) prompted us to investigate other isolates for this activity. Three other isolates from clade D2 (6LA4, 3LE1 and 2LD2) also exhibited activity against P. vulgaris, but at levels below 70 % (40–61 %), thus they were not indicated as active in Table 2. Extracts from 5LA7, 6LA4 and 3LE1 fermented in BFM4 were examined by UPLC-HRMS/ELSD/PDA. Four major peaks were present in the ELSD chromatograms and corresponded to the following ions (m/z): 255.0656, 271.0606, 429.1188 and 912.6238 amu. Based on searching AntiBase 2012, the ions with m/z 255 and 271 amu may correspond to daidzein and other related isoflavones, although multiple matches were observed for these masses, thus the identification is tentative. The remaining two ions were purified (data not shown) and identified as the known compounds 5-deoxyenterocin (m/z 429 amu) and reginamide A (m/z 912 amu) by comparison of their MS and NMR spectral data with those reported in the literature [52, 72]. Both compounds were tested for cytotoxicity against the two immortalized cell lines and four microorganisms at 50 μg mL−1. Reginamide A, the major compound in extracts of the three strains, showed weak activity (34 % inhibition) against VRE, while 5-deoxyenterocin exhibited similarly weak activity (37 %) against MRSA. The putative isoflavones (m/z 255 and 271 amu) were present in BFM4 fermentation extracts and may be partially responsible for the observed activity, however, these ions were absent in other active extracts of fermentations performed in other media (BFM1-3), suggesting that these metabolites were not responsible for the observed activity. It is possible that the observed P. vulgaris activity is due to the combined effects of two or more of the ions described above, or that a metabolite(s) other than the four major peaks are responsible for the activity. Bioassay guided fractionation would be necessary to identify the metabolite(s) responsible for the observed activity.
Identification of cytotoxic metabolites from Actinoalloteichus sp. 2L868
Due to the potent cytotoxic activity exhibited by Actinoalloteichus sp. 2L868 extracts, large scale fermentation was conducted and cytotoxic metabolites identified by bioassay-guided fractionation. The cytotoxic activity of fractions and purified compounds was assessed against immortalized BJ and HTB-26 cell lines. The crude extract of the large scale fermentation was first tested against the immortalized cell lines to confirm activity comparable to that observed in primary screening against the primary HMT3909S8 cell line. Bioassay-guided fractionation led to the isolation of inseparable mixtures of cytotoxic saturated fatty acids (FAs), saturated monohydroxylated FAs, polyhydroxylated unsaturated FAs, glycerolipids (hydroxylated FAs) and polyunsaturated monohydroxylated FAs (Table S6). 1-Isopentadecanoyl-3β-d-glucopyranosyl-X-glycerol [65] was purified to homogeneity and almost completely inhibited the growth of HTB26 cells (91 %) at a 20 μg mL−1, while BJ fibroblasts were unaffected by an identical concentration.
Metabolomic screening of actinomycete fermentation extracts
To explore the metabolomes of actinomycete isolates from NL sediments, 28 isolates were subjected to UPLC-HRMS-based metabolomics analysis (Table 2, MET column). 2L868, 6LA4, 3LE1 and 5LA7 were not included in the metabolomics analysis as these strains were subjected to in-depth manual chemical analysis due to their observed cytotoxic and P. vulgaris activity (above). After removal of buckets (defined by a m/z ± 0.01 amu and retention time ± 0.1 min) [23] detected in methanol blanks and ISP2 media blanks the metabolomics data set was composed of 606 buckets with mass ratios ranging from m/z 192.0664–1699.8929 amu and retention times ranging from 0.39 to 8.54 min. The majority of buckets (82.5 %) were only observed in a single isolate, while a small number of buckets were widely distributed; only 10 buckets were observed in five or more strains and the maximum number of strains a single bucket was detected in was 10. Collectively, these results suggest a high degree of chemical diversity in the metabolomes of the actinomycetes analyzed. The number of buckets varied greatly between strains with the lowest number of buckets detected in 8LC4 (2) and the most detected in 8LD1 (170). To explore the relationships between actinomycete metabolic profiles a cluster analysis was performed (Fig. 3). As observed previously, the cluster analysis was correlated with chemical diversity, with isolates producing the greatest number of buckets at the top of the cladogram and those producing the fewest at the bottom [23]. In general, strains clustered independently of phylogenetic clade or OTU assignment (Table 2; Fig. 2). For example, representative strains from the same OTU (7LC12/3LA9 − OTU1; 5LB9/8LD8 − OTU18, Table 2) did not cluster together in Fig. 3. In both cases, the strains from the same OTU did not produced common metabolites. Conversely, strains from different OTUs, which belonged to different clades in Fig. 2, exhibited multiple shared metabolites. For example, the metabolic profiles of 8LC9, 3LD3 and 1LA4, which belonged to clades D2, E2 and E3 in Fig. 2, contained 12 shared metabolites. The shared metabolites accounted for 37.5–46.2 % of the total buckets identified in each of these strains. Similar results were observed for the following clades in the metabolomics cluster analysis: 1LC9/2LD2 (11 shared buckets), 5LB7/8LC5/2LA10 (8 shared buckets), and 7LA3/5LA1 (7 shared buckets) (Fig. 3). Interestingly, antimicrobial activity appeared to be associated with particular clades within the metabolomics cluster analysis. For instance, 8LC9, 3LD3 and 1LA4 clustered together in Fig. 3, sharing 12 buckets in common, and extracts from these organisms all exhibited activity against C. albicans. No buckets were shared between these three strains and the other three strains which inhibited the growth of C. albicans. Similarly, two (1LC9 and 2LD2) of three strains exhibiting activity against MRSA formed a cluster in the metabolomics analysis and had 11 buckets in common. We did not attempt to identify the bioactive metabolites from the members of the two previously mentioned clades, thus we cannot definitively attribute the bioactivity to buckets common to all members of each clade. However, these observations suggest that metabolomics analysis is able to not only identify strains with similar chemical profiles but can also be used to predict strains likely to exhibit similar bioactivity profiles.

Metabolomics-based cluster analysis of 18 actinomycetes
To further explore the metabolic potential of the actinomycetes isolated from NL we subjected the transformed UPLC-HRMS data to principle component analysis (PCA) to identify chemical signatures unique to the data set. PCA identified three strains with chemical signatures unique among the 28 strains analyzed. Based on comparison of HRMS and UV spectra to published data, the compounds responsible for differentiating the chemical profiles of the 7LC12 and 8LD1 extracts from those of the other actinomycetes examined were identified as arylomycins [66] and nactins [14], respectively. Arylomycins are inhibitors of bacterial type 1 signal peptidase (SPase) and have reported broad-spectrum antimicrobial activity [73]. Despite the presence of arylomycins in the extract of 7LC12, no antimicrobial activity was detected. This lack of activity is likely due to mutations present in SPase, which render target bacteria resistant to arylomycins [73]. Conversely, the nactin family of cyclic ionophores is known to possess antimicrobial activity against Gram-positive bacteria and fungi; nactins are presumably responsible for the observed antimicrobial activity exhibited by 8LD1 extracts [14]. PCA also flagged the 8LB7 extract as containing metabolites unique to the metabolomic data set. UPLC-HRMS analysis of the 8LB7 extract identified the valinomycins [9] and the identity of these compounds was confirmed by tandem mass spectrometry (data not shown). The 8LB7 extract also contained an ion with a m/z of 1,592.8184 amu. The metabolite was purified and the structure determined by NMR spectroscopy and HRMS. At the time of isolation the compound was novel, however, Um and colleagues [78] recently reported the identical structure of the lasso peptide sungsanpin. Sungsanpin was tested for antimicrobial activity and was inactive. Valinomycin did not exhibit antibacterial activity against the test organisms utilized in this study, but has been reported to inhibit the growth of C. albicans with a minimum inhibitory concentration of approximately, 12 μg mL−1 [58, 69]. The lack of C. albicans activity by the 8LB7 extract is likely due to an insufficient concentration of valinomycins in the extract.
Discussion
Actinobacterial diversity
Culture-independent approaches to assessing actinobacterial diversity in environmental samples typically rely on the amplification of Actinobacteria 16S rDNA gene fragments using PCR primers designed to selectively amplify actinobacterial sequences [75]. The resulting amplicons are cloned, dereplicated and then a representative subset of the clones sequenced. Due to the labor intensive nature of clone library generation usually only a modest number of clones are sequenced, and at most a few hundred sequences are obtained per sample. The use of Actinobacteria-specific primers are needed when assessing actinobacterial diversity using clone libraries to avoid actinobacterial sequences from being obfuscated by sequences from more abundant taxa [75]. Amplicon pyrosequencing generates large quantities of sequence data without the need for cloning, thereby allowing for in-depth assessment of bacterial community composition without the bias of culture based methods [13, 20]. This study did not employ Actinobacteria-targeted primers; instead we used non-selective primers and relied on the superior sequencing depth of pyrosequencing to uncover actinobacterial diversity within the background of overall bacterial diversity. Despite the generation of thousands of sequences per sample a limited number of actinobacterial sequences were obtained. The sequencing depth was sufficient to uncover a high level of overall bacterial diversity as evidenced by H′ values exceeding 6 for all but one sediment and coverage values ranging from 64 to 85 %. In contrast, the sampling depth of actinobacterial sequences was clearly insufficient to comprehensively describe actinobacterial diversity as evidenced by relatively low coverage values for 5 out of 8 sediments (Table 1) and rarefaction curves which were far from reaching their asymptote (Fig. S1). Despite the relatively low sampling coverage, significant actinobacterial diversity was calculated for the NL sediments, indicated by H′ values ranging from 2.45 to 3.76 and richness estimates ranging from 29 to 110 OTUs. This level of actinobacterial diversity is comparable to diversity reported from studies utilizing Actinobacteria-specific primers to elucidate actinobacterial diversity in corals and marine sediments [62, 86, 87]. An advantage to using non-selective primers to assess bacterial diversity is that it allows one to assess abundance of Actinobacteria relative to other bacterial groups in a given habitat. The prevalence of Actinobacteria in NL sediments (<6 %) is similar to the prevalence observed in geographically diverse marine sediments, which varies from rare to <8 % [21, 26, 38, 79]. In comparison, Actinobacteria may comprise up to 30 % of soil microbial communities [1, 40]. This difference suggests that Actinobacteria may play a smaller role in nutrient cycling in sediments than in soils. Interestingly, sequence libraries from NL were devoid of the filamentous, spore-forming Actinobacteria most commonly isolated from marine sediments, namely Streptomyces and Micromonospora. A similar underrepresentation of these taxa was also observed in marine sediments from New Brunswick (NB), Canada [21] and has been noted in several studies, including those employing Actinobacteria-specific primer sets [4, 86, 87]. Indeed, detection of these groups often requires the use of family or genus-specific primers [4, 62]. The lack of detection of these bacteria via pyrosequencing may be a result of insufficient sequencing depth, preventing the detection of low abundance OTUs, which can readily be cultured when highly selective isolation techniques are applied. Furthermore, these taxa may exist in marine sediments as spores, which are particularly resilient to cell lysis and therefore, may be underrepresented in environmental DNA [35]. Future studies should explore the use of Actinobacteria-specific primers in conjunction with the sequencing depth afforded by pyrosequencing to more fully explore the extent of Actinobacteria in natural habitats.
A highly selective isolation strategy was utilized to assess the phylogenetic diversity of filamentous actinomycetes present in NL sediments, as previous studies of marine sediments have indicated that filamentous actinomycetes are rare members in these habitats [21, 62]. Application of such an approach was supported by culture-independent analysis of NL sediments which indicated Actinobacteria comprised a small component (<6 %) of the extant bacterial diversity present in these sediments. Over 1,500 actinomycete were isolated from NL sediments and 360 characterized by partial 16S rDNA sequencing. The taxonomic diversity of filamentous actinomycetes isolated from NL sediments was limited to Promicromonospora, Nocardiopsis, Actinoalloteichus and Streptomyces-related isolates. Sequences matching those of the cultured isolates were not found in the culture-independent sequence libraries, underscoring the complimentary nature of culture-dependent and culture-independent studies in investigations of bacterial diversity [70]. A nearly identical distribution of genera was obtained from NB sediments with the exception of Actinoalloteichus species, which were not isolated from NB sediments. In comparison to other studies of culturable actinomycete diversity, the taxonomic diversity of filamentous actinomycetes obtained from Atlantic Canadian sediments was somewhat lower. In part, the lower diversity of genera can be explained by the fact that unicellular actinomycetes commonly isolated from marine environments, such as Micrococcaceae, Microbacteriaceae and Dermacoccaceae [29, 60], would not have been isolated in our study due to the focus on filamentous actinomycetes. Interestingly, Micromonospora isolates were not obtained from NB or NL sediments despite the fact that they have been reported to be readily cultured from marine habitats [8, 30]. The reason for this is most likely due to differences in isolation procedures [8]. The diversity of Micromonospora and other so called “rare actinomycetes” may not have been thoroughly investigated using the isolation approaches employed in this study. To further explore the culturable diversity of these interesting actinomycetes in sediments from Atlantic Canada highly focused isolation techniques need to be applied.
To compare the diversity of actinomycetes obtained from different NL sediments (360) a NJ tree was constructed (Fig. 2). Clades containing different genera were well supported with bootstrap values >90 %, however, nodes within the major Streptomyces clades were supported by low bootstrap values. Consequently, it is difficult to draw conclusions regarding the biogeographic distribution of streptomycetes in NL sediments. The lack of resolution within the tree is likely due to the highly related nature of the 16S rRNA gene within the genus Streptomyces and the partial sequences used for constructing the tree. Examining the distribution of isolates among the 59 OTUs (Table S4) indicates that the majority of isolates (90.5 %) are widely distributed in NL sediments (e.g. belong to OTUs consisting of multiple isolates from multiple locations, Table S4), and is consistent with the generally ubiquitous distribution of actinomycetes in natural environments [84]. Conversely, singleton OTUs account for 9.5 % of isolates and may represent “rare” Streptomyces phylotypes which may only be obtained by investigating geographically diverse samples. Further sampling and the application of higher resolution approaches, such as multi-locus sequence typing, would be required to fully explore the biogeographic distribution of streptomycetes in NL sediments [19, 25].
Metabolic potential of NL actinomycetes
The 59 actinomycete phylotypes isolated from NL sediments provided an exciting resource to explore for bioactive secondary metabolites [59, 80]. Two complimentary approaches were taken to screen selected isolates for secondary metabolite production. Firstly, fermentation extracts were screened for anticancer (cytotoxic) and antimicrobial activity. Secondly, fermentation extracts were screened for the presence of novel metabolites using a metabolomics approach [23].
Forty-one percent of isolates exhibited antimicrobial activity in one or more bioassays with the majority of activity exhibited against C. albicans (31 %), followed by the two Gram-positive pathogens (25 %). Only a single isolate exhibited >70 % growth inhibition against P. vulgaris, although two closely related strains also exhibited weak activity against this pathogen. Similar distributions of bioactivity have been observed in other studies, where activity against yeast and Gram-positive bacteria is observed most frequently, while activity against Gram-negative bacteria is rare [8, 30, 55]. While only a small number of strains were tested for anticancer activity, a relatively high percentage (33 %) exhibited activity. Primary breast cancer cells were used in the primary screen because primary cell lines have been shown to more closely resemble tumors encountered in clinical settings; therefore they provide a more reliable model of in vivo anticancer activity than traditional immortalized laboratory cell lines [57]. Due to the taxonomic novelty of Actinoalloteichus sp. 2L868 and the strong cytotoxic activity exhibited by extracts from this strain, a bioassay-guided fractionation approach was taken to isolate the active metabolites produced by this organism. The bioactivity was associated with non-polar fractions containing inseparable mixtures of fatty acids, many of which exhibited strong cytotoxic activity at a moderate concentration (20 μg mL−1). Interestingly, 1-Isopentadecanoyl-3β-d-glucopyranosyl-X-glycerol was also purified from the strain and shown to exhibit selective cytotoxicity against breast cancer cells. This glycolipid was previously reported from the myxobacterium Cystobacter fuscus, and to the best of our knowledge, this is the first report of the biological activity of this metabolite and the first report of its production by an actinomycete [65]. The bioactivities exhibited by the fatty acids produced by 2L868 are consistent with cytotoxic activities previously reported for this family of compounds [6, 15, 49, 71].
Within the scope of NP discovery programs metabolomic screening is useful for both the dereplication of isolates with similar metabolic profiles and for the discovery of strains producing novel metabolites [23, 27, 42]. The study of 28 actinomycetes demonstrated the ability of metabolomics to identify strains producing similar suites of compounds based on grouping in cluster analysis of the metabolomic data set. The identification of groups with similar chemical profiles would not be easily predicted based on a taxonomic analysis alone, as the isolates producing similar chemical profiles were not closely related phylogenetically. For instance, 8LC9, 3LD3 and 1LA4 exhibited similar metabolomic profiles (Fig. 3) but belonged to different clades in the phylogenetic analysis, which in the case of clades D2 and E2/E3 (Fig. 2) were very well supported by bootstrap analysis. Similarly, strains which belonged to the same OTU did not contain significant overlap in their metabolic profiles. These observations are consistent with previous comparisons of cluster analyses performed on metabolomic and 16S rDNA sequence data sets, and highlight the benefits of metabolomic versus 16S rDNA sequence-based dereplication [23]. Metabolomics-based discovery of NPs also offers the advantage that when coupled with appropriate statistical analyses (e.g. PCA), unique metabolites can be readily identified irrespective of observable biological activity. The value of incorporating metabolomics screening in NP discovery programs was illustrated in this study by the identification of two metabolites, arylomycin and sungsanpin, which would not have been identified utilizing a discovery paradigm based solely on identifying bioactive extracts. Potentially interesting metabolites may be overlooked in activity-oriented NP screening programs for several reasons: they may not be present at a concentration necessary to evoke a biological response, interference from other components of complex NP mixtures may mask their activity, or the assay system may simply not be able to detect all potential inhibitors of the targeted process due to intrinsic limitations. For example, arylomycins, which are of great interest due to their unique structure and the novelty of their biological target (SPase), were not identified in our whole cell antimicrobial assay as the test organisms utilized in this study are naturally resistant due to mutations in SPase [73]. Without the inclusion of complimentary metabolomic screening this promising antibiotic would have otherwise been completely overlooked.
Conclusion
Small subunit rRNA amplicon pyrosequencing is powerful approach to characterizing extant bacterial diversity in environmental samples without the biases inherent in culture-based approaches [32]. Using such an approach, NL sediments were demonstrated to host diverse overall bacterial communities, although estimates of sampling coverage indicated significant actinobacterial diversity remained to be discovered. Undoubtedly, future research combining Actinobacteria-specific primers with pyrosequencing will uncover unprecedented levels of actinobacterial diversity and provide a more comprehensive assessment of actinobacterial communities in natural habitats. Culture-dependent analysis indicated that Streptomyces were the most abundant group of cultivable actinomycetes in NL sediments while Nocardiopsis, Actinoalloteichus and Promicromonospora species constituted rare members of the culturable actinomycete community. No sequences of cultured actinomycetes were detected in the culture-independent analysis, underscoring the complimentary nature of culture-dependent and culture–independent approaches when attempting to describe bacterial diversity [69]. A total of 59 OTUs were identified from 360 actinomycete isolates, indicating a significant level of species-level actinomycete diversity in NL sediments. As the majority of interest in actinomycetes stems from their ability to produce bioactive metabolites [5], we assessed the metabolic potential of a representative set of isolates using a combination of bioassays and metabolomics screening. Despite screening only a small percentage of the actinomycete diversity cultivated from NL sediments under a limited set of fermentation conditions, high levels of biological activity were detected (40 % of all strains) and several rare metabolites were identified. These results suggest that comprehensive screening of the remaining 328 actinomycetes isolated from NL sediments will uncover significant additional, and potentially novel, chemical diversity. The effectiveness of metabolomics-based strain dereplication was confirmed in this study and the value of metabolomics screening to NP discovery efforts was validated by the identification of two rare secondary metabolites that would not have been discovered via standard biological screening. These results argue strongly for the continued development of metabolomics screening methodologies and their further integration in NP discovery programs. Future coupling of metabolomics discovery work flows with novel strain prioritization approaches [85] will further invigorate NP discovery pipelines.
References
Acosta-Martínez V, Dowd S, Sun Y, Allen V (2008) Tag-encoded pyrosequencing analysis of bacterial diversity in a single soil type as affected by management and land use. Soil Biol Biochem 40:2762–2770
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ (1990) Basic local alignment search tool. J Mol Biol 1990:403–410
Atlas RM, Parks LC (1993) Handbook of microbiological media. CRC Press, Boca Raton
Babalola OO, Kirby BM, Le Roes-Hill M, Cook AE, Cary SC, Burton SG, Cowan DA (2009) Phylogenetic analysis of actinobacterial populations associated with AntarcticDry Valley mineral soils. Environ Microbiol 11:566–576
Bérdy J (2005) Bioactive microbial metabolites. J Antibiot 58:1–26
Berquin IM, Edwards IJ, Chen YQ (2008) Multi-targeted therapy of cancer by omega-3 fatty acids. Cancer Lett 269:363–377
Blunt JW, Copp BR, Munro MH, Northcote PT, Prinsep MR (2010) Marine natural products. Nat Prod Rep 27:165–237
Bredholt H, Fjaervik E, Johnsen G, Zotchev SB (2008) Actinomycetes from sediments in the Trondheim fjord, Norway: diversity and biological activity. Mar Drugs 6:12–24
Brockmann H, Schmidt-Kastner G (1955) Valinomycin I, XXVII. Mitteilung über Antibiotika aus Actinomyceten. Chem Ber 88:57–61
Bull AT, Stach JE (2007) Marine actinobacteria: new opportunities for natural product search and discovery. Trends Microbiol 15:491–499
Cermeño P, Falkowski PG (2009) Controls on diatom biogeography in the ocean. Science 325:1539–1541
Chao A, Lee S (1992) Estimating the number of classes via sample coverage. J Am Stat Assoc 87:210–217
Chu H, Fierer N, Lauber CL, Caporaso JG, Knight R, Grogan P (2010) Soil bacterial diversity in the Arctic is not fundamentally different from that found in other biomes. Environ Microbiol 12:2998–3006
Corbaz R, Ettlinger L, Gaumann E, Keller-Schierlein K, Kradolfer F, Neipp L, Prelog V, Zahner H (1955) Stoffwechselprodukte con Actinomyceten. 3. Mitteileng Nonactin Helv Chim Acta 38:1445–1448
Das UN (1992) Anti-cancer effects of cis-unsaturated fatty acids both in vitro and in vivo. In: Lipid-soluble antioxidants: biochemistry and clinical applications, p 482
de Wit R, Bouvier T (2006) ‘Everything is everywhere, but, the environment selects’; what did Baas Becking and Beijerinck really say? Environ Microbiol 8:755–758
DeLong EF (1997) Marine microbial diversity: the tip of the iceberg. Trends Biotechnol 15:203–207
Demain AL, Sanchez S (2009) Microbial drug discovery: 80 years of progress. J Antibiot (Tokyo) 62:5–16
Doroghazi JR, Buckley DH (2010) Widespread homologous recombination within and between Streptomyces species. ISME J 4:1136–1143
Dowd SE, Callaway TR, Wolcott RD, Sun Y, McKeehan T, Hagevoort RG, Edrington TS (2008) Evaluation of the bacterial diversity in the feces of cattle using 16S rDNA bacterial tag-encoded FLX amplicon pyrosequencing (bTEFAP). BCM Microbiol 8:125
Duncan K, Haltli B, Gill KA, Kerr RG (2014) Bioprospecting from marine sediments of New Brunswuck, Canada: exploring the relationship between total bacterial diversity and actinobacteria diversity. Mar Drugs 12:899–925
Feling RH, Buchanan GO, Mincer TJ, Kauffman CA, Jensen PR, Fenical W (2003) Salinosporamide A: a highly cytotoxic proteasome inhibitor from a novel microbial source, a marine bacterium of the new genus Salinospora. Angew Chem Int Ed Engl 42:355–357
Forner D, Berrué F, Correa H, Duncan K, Kerr RG (2013) Chemical dereplication of marine actinomycetes by liquid chromatography-high resolution mass spectrometry profiling and statistical analysis. Anal Chim Acta 805:70–79
Freel KC, Edlund A, Jensen PR (2012) Microdiversity and evidence for high dispersal rates in the marine actinomycete ‘Salinispora pacifica’. Environ Microbiol 14:480–493
Freel KC, Millán-Aguiñaga N, Jensen PR (2013) Multilocus sequence typing reveals evidence of homologous recombination linked to antibiotic resistance in the genus Salinispora. Appl Environ Microbiol 79:5997–6005
Gao F, Li F, Tan J, Yan J, Sun H (2014) Bacterial community composition in the gut content and ambient sediment of sea cucumber Apostichopus japonicus revealed by16S rRNA gene pyrosequencing. PLoS One 9:e100092
Gill KA, Berrué F, Arens JC, Kerr RG (2014) Isolation and structure elucidation of cystargamide, a lipopeptide from Kitasatospora cystarginea. J Nat Prod 77:1372–13766
Giovannoni SJ, Stingl U (2005) Molecular diversity and ecology of microbial plankton. Nature 437:343–348
Gontang EA, Fenical W, Jensen PR (2007) Phylogenetic diversity of gram-positive bacteria cultured from marine sediments. Appl Environ Microbiol 73:3272–3282
Hong K, Gao A, Xie Q, Gao H, Zhuang L, Lin H, Yu H, Li J, Yao X, Goodfellow M, Ruan J (2009) Actinomycetes for marine drug discovery isolated from mangrove soils and plants in China. Mar Drugs 7:24–44
Hubert C et al (2009) A constant flux of diverse thermophilic bacteria into the cold Arctic seabed. Science 325:1541–1544
Hugenholtz P, Goebel BM, Pace NR (1998) Impact of culture-independent studies on the emerging phylogenetic view of bacterial diversity. J Bacteriol 180:4765
Jensen PR, Dwight R, Fenical W (1991) Distribution of actinomycetes in near-shore tropical marine sediments. Appl Environ Microbiol 57:1102–1108
Jensen PR, Gontang E, Mafnas C, Mincer TJ, Fenical W (2005) Culturable marine actinomycete diversity from tropical Pacific Ocean sediments. Environ Microbiol 7:1039–1048
Jensen PR, Lauro FM (2008) An assessment of actinobacterial diversity in the marine environment. Antonie Van Leeuwenhoek 94:51–62
Jensen PR, Mafnas C (2006) Biogeography of the marine actinomycete Salinispora. Environ Microbiol 8:1881–1888
Kaeberlein T, Lewis K, Epstein SS (2002) Isolating “uncultivable” microorganisms in pure culture in a simulated natural environment. Science 296:1127–1129
Kerfahi D, Hall-Spencer JM, Tripathi BM, Milazzo M, Lee J, Adams JM (2014) Shallow water marine sediment bacterial community shifts along a natural CO2 gradient in the Mediterranean Sea off Vulcano, Italy. Microb Ecol 67:819–828
Kieser T, Bibb MJ, Buttner MJ, Chater KF, Hopwood DA (2000) Practical Streptomyces manual. The John Innes Foundation, Norwich
Kirchman DL, Cottrell MT, Lovejoy C (2010) The structure of bacterial communities in the western Arctic Ocean as revealed by pyrosequencing of 16S rRNA genes. Environ Microbiol 12:1132–1143
Koehn FE, Carter GT (2005) Rediscovering natural products as a source of new drugs. Discov Med 5:159–164
Krug D, Müller R (2014) Secondary metabolomics: the impact of mass spectrometry-based approaches on the discovery and characterization of microbial natural products. Nat Prod Rep 31:768–783
Lebar MD, Heimbegner JL, Baker BJ (2007) Cold-water marine natural products. Nat Prod Rep 24:774–797
Liu X, Ashforth E, Ren B, Song F, Dai H, Liu M, Wang J, Xie Q, Zhang L (2010) Bioprospecting microbial natural product libraries from the marine environment for drug discovery. J Antibiot 63:415–422
Maldonado LA, Fragoso-Yáñez D, Pérez-García A, Rosellón-Druker J, Quintana ET (2009) Actinobacterial diversity from marine sediments collected in Mexico. Antonie Van Leeuwenhoek 95:111–120
Maldonado LA, Stach JE, Pathom-aree W, Ward AC, Bull AT, Goodfellow M (2005) Diversity of cultivable actinobacteria in geographically widespread marine sediments. Antonie Van Leeuwenhoek 87:11–18
Manam RR et al (2005) Lajollamycin, a nitro-tetraene spiro-beta-lactone-gamma-lactam antibiotic from the marine actinomycete Streptomyces nodosus. J Nat Prod 68:240–243
Martiny JB et al (2006) Microbial biogeography: putting microorganisms on the map. Nat Rev Microbiol 4:102–112
Menendez JA et al (2001) Effects of gamma-linolenic acid and oleic acid on paclitaxel cytotoxicity in human breast cancer cells. Eur J Cancer 37:402–413
Mincer TJ, Fenical W, Jensen PR (2005) Culture-dependent and culture-independent diversity within the obligate marine actinomycete genus Salinispora. Appl Environ Microbiol 71:7019–7028
Mincer TJ, Jensen PR, Kauffman CA, Fenical W (2002) Widespread and persistent populations of a major new marine actinomycete taxon in ocean sediments. Appl Environ Microbiol 68:5005–5011
Mohimani H, Liu WT, Yang YL, Gaudêncio SP, Fenical W, Dorrestein PC, Pevzner PA (2011) Multiplex de novo sequencing of peptide antibiotics. J Comput Biol 18:1371–1381
Montalvo NF, Mohamed NM, Enticknap JJ, Hill RT (2005) Novel actinobacteria from marine sponges. Antonie Van Leeuwenhoek 87:29–36
Nacke H, Thürmer A, Wollherr A, Will C, Hodac L, Herold N, Schöning I, Schrumpf M, Daniel R (2011) Pyrosequencing-based assessment of bacterial community structure along different management types in German forest and grassland soils. PLoS One 16:e17000
Nakashima T, Anzai K, Suzuki R, Kuwahara N, Takeshita S, Kanamoto A, Ando K (2009) Productivity of bioactive compounds in Streptomyces species isolated from Nagasaki marine environments. Actinomycetologica 23:16–20
National Committee for Clinical Laboratory Standards (2003) Methods for dilution antimicrobial susceptibility tests for bacteria that grow aerobically: approved standard—sixth edition. NCCLS document M7-A6. National Committee for Clinical Laboratory Standards, Wayne, Pa
Neve RM et al (2006) A collection of breast cancer cell lines for the study of functionally distinct cancer subtypes. Cancer Cell 10:515–527
Okoli I, Coleman JJ, Tampakakis E, An WF, Holson E, Wagner F, Conery AL, Larkins-Ford J, Wu G, Stern A, Ausubel FM, Mylonakis E (2009) Identification of antifungal compounds active against Candida albicans using an improved high-throughput Caenorhabditis elegans assay. PLoS One 4:e7025
Olano C, Méndez C, Salas JA (2009) Antitumor compounds from marine actinomycetes. Mar Drugs 7:210–248
Pathom-Aree W, Stach JE, Ward AC, Horikoshi K, Bull AT, Goodfellow M (2006) Diversity of actinomycetes isolated from Challenger Deep sediment (10,898 m) from the Mariana Trench. Extremophiles 10:181–189
Pluskal T, Castillo S, Villar-Briones A, Oresic M (2010) MZmine 2: modular framework for processing, visualizing, and analyzing mass spectrometry-based molecular profile data. BMC Bioinform 11:395
Prieto-Davó A, Villarreal-Gómez LJ, Forschner-Dancause S, Bull AT, Stach JE, Smith DC, Rowley DC, Jensen PR (2013) Targeted search for actinomycetes from nearshore and deep-sea marine sediments. FEMS Microbiol Ecol 84:510–518
Ramette A, Tiedje JM (2007) Multiscale responses of microbial life to spatial distance and environmental heterogeneity in a patchy ecosystem. Proc Natl Acad Sci USA 104:2761–2766
Riedlinger J et al (2004) Abyssomicins, inhibitors of the para-aminobenzoic acid pathway produced by the marine Verrucosispora strain AB-18-032. J Antibiot 57:271–279
Scherer OW, Budzikiewicz H, Hartmann R, Klein RA, Egge H (1992) The structural elucidation of the two positional isomers of a monoglucopyranosyl monoacyl glycerol derivative from Cystobacter fuscus (Myxobacterales). Biochim Biophys Acta 1117:42–46
Schimana J, Gebhardt K, Holtzel A, Schmid DG, Sussmuth R, Muller J, Pukall R, Fiedler HP (2002) Arylomycins A and B, new biaryl-bridged lipopeptide antibiotics produced by Streptomyces sp. Tü 6075. I. Taxonomy, fermentation, isolation and biological activities. J Antibiot 56:565–570
Schloss PD Mothur wiki SOP: http://www.mothur.org/wiki/454_SOP. Accessed Feb, 2013
Schloss PD, Gevers D, Westcott SL (2011) Reducing the effects of PCR amplification and sequencing artifacts on 16S rRNA-based studies. PLoS One 6:e27310
Seshachalam D, Frahm DH, Ferraro FM (1973) Cation reversal of inhibition of growth by valinomycin in Streptococcus pyogenes and Clostridium sporogenes. Antimicrob Agents Chemother 3:63–67
Shade A, Hogan CS, Klimowicz AK, Linske M, McManus PS, Handelsman J (2012) Culturing captures members of soil rare biosphere. Environ Microbiol 14:2247–2252
Shaikh IA, Brown I, Wahle KW, Heys SD (2010) Enhancing cytotoxic therapies for breast and prostate cancers with polyunsaturated fatty acids. Nutr Cancer 62:284–296
Sitachitta N, GadepalliM Davidson BS (1996) New a-pyrone-containing metabolites from a marine-derived actinomycete. Tetrahedron 52:8073–8080
Smith PA, Roberts TC, Romesberg FE (2010) Broad spectrum antibiotic activity of the arylomycin natural products is masked by natural target mutations. Chem Biol 17:1223–1231
Sogin ML, Morrison HG, Huber JA, Mark Welch D, Huse SM, Neal PR, Arrieta JM, Herndl GJ (2006) Microbial diversity in the deep sea and the underexplored “rare biosphere”. Proc Natl Acad Sci USA 103:12115–12120
Stach JE, Maldonado LA, Masson DG, Ward AC, Goodfellow M, Bull AT (2003) Statistical approaches for estimating actinobacterial diversity in marine sediments. Appl Environ Microbiol 69:6189–6200
Sunagawa S, Woodley CM, Medina M (2010) Threatened corals provide underexplored microbial habitats. PLoS One 5:e9554
Takahashi Y, Omura S (2003) Isolation of new actinomycete strains for the screening of new bioactive compounds. J Gen Appl Microbiol 49:141–152
Um S, Kim YJ, Kwon H, Wen H, Kim SH, Kwon HC, Park S, Shin J, Oh DC (2013) Sungsanpin, a lasso peptide from a deep-sea streptomycete. J Nat Prod 76:873–879
Wang Y, Sheng HF, He Y, Wu JY, Jiang YX, Tam NF, Zhou HW (2012) Comparison of the levels of bacterial diversity in freshwater, intertidal wetland, and marine sediments by using millions of illumina tags. Appl Environ Microbiol 78:8264–8271
Watve MG, Tickoo R, Jog MM, Bhole BD (2001) How many antibiotics are produced by the genus Streptomyces? Arch Microbiol 176:386–390
Weisburg WG, Barns SM, Pelletier DA, Lane DJ (1991) 16S ribosomal DNA amplification for phylogenetic study. J Bacteriol 173:697–703
Weyland H (1969) Actinomycetes in North Sea and Atlantic Ocean sediments. Nature 223:858
Whitaker RJ, Grogan DW, Taylor JW (2003) Geographic barriers isolate endemic populations of hyperthermophilic archaea. Science 301:976–978
Wu J, Guan T, Jiang H, Zhi X, Tang S, Dong H, Zhang L, Li W (2009) vDiversity of Actinobacterial community in saline sediments from Yunnan and Xinjiang, China. Extremophiles 13:623–632
Xie P, Ma M, Rateb ME, Shaaban KA, Yu Z, Huang SX, Zhao LX, Zhu X, Yan Y, Peterson RM, Lohman JR, Yang D, Yin M, Rudolf JD, Jiang Y, Duan Y, Shen B (2014) Biosynthetic potential-based strain prioritization for natural product discovery: a showcase for diterpenoid-producing actinomycetes. J Nat Prod 77:377–387
Yang S, Sun W, Tang C, Jin L, Zhang F, Li Z (2013) Phylogenetic diversity of actinobacteria associated with soft coral Alcyonium gracllimum and stony coral Tubastraea coccinea in the East China Sea. Microb Ecol 66:189–199
Zhang G, Cao T, Ying J, Yang Y, Ma L (2014) Diversity and novelty of actinobacteria in Arctic marine sediments. Antonie Van Leeuwenhoek 105:743–754
Acknowledgments
The authors would like to acknowledge C. Wilde (AvantiCell Science, Ayr, Scotland) for primary breast cancer cell line cytotoxicity screening, N. Duncan (Nautilus Biosciences Canada Inc.) for assistance with fermentations and extractions and M. Lanteigne (UPEI) for conducting antimicrobial assays and in-house cytotoxicity assays. Financial support was received from the Natural Sciences and Engineering Council of Canada, the Canada Research Chair program, the Atlantic Innovation Fund and the Jeanne and Jean-Louis Lévesque Foundation. Katherine Duncan gratefully acknowledges financial support from the Thunder Cove Breast Cancer Researcher Award and a PhD Fellowship from the PEI Innovation Foundation.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Duncan, K.R., Haltli, B., Gill, K.A. et al. Exploring the diversity and metabolic potential of actinomycetes from temperate marine sediments from Newfoundland, Canada. J Ind Microbiol Biotechnol 42, 57–72 (2015). https://doi.org/10.1007/s10295-014-1529-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10295-014-1529-x