
Abstract
The metagenomic approach has greatly accelerated the discovery of new enzymes by giving access to the genetic potential of microorganisms from various environments. Function-based screening depends on adequate expression of the foreign genes in the heterologous host, which can be challenging in large-insert libraries. In this study, the shuttle cosmid vector pFX583 was used for the construction and screening of a metagenomic library. This vector allows T7 RNA polymerase-directed transcription of the cloned DNA and can be used in Escherichia coli and Streptomyces lividans. The DNA used for the library construction was obtained from an enriched biomass. The library was screened for lipolytic and proteolytic activities using E. coli and S. lividans as hosts. Numerous E. coli clones with lipolytic activity were detected. Unfortunately, proteases could not be detected in both hosts. From the lipolytic activity screen, a gene coding for a new lipase was isolated, and partial characterization was conducted.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Considering that nearly 99% of the microorganisms in nature are not readily culturable [1], a large genetic reservoir remains untapped despite years of culture-dependent screening studies. To gain access to this genetic information, methods were developed based on the analysis and direct isolation of nucleic acids from uncultured microorganisms. Among those methods, metagenomics, the analysis of the collective microbial genomes present in a given habitat, has emerged as a powerful approach [37]. Metagenomics usually involves direct isolation of genomic DNA from an environment, construction of a library by cloning the DNA into a suitable vector, and subsequent high-throughput sequencing or screening. Screens of metagenomic libraries can be based on either sequence- or function-driven approaches. Sequence-based screening relies on the use of a conserved DNA sequence for designing hybridization probes or polymerase chain reaction (PCR) primers to detect specific sequences [35]. The main advantage of sequence-driven screening methods is their independence toward expression of the cloned gene by the heterologous host. However, it tends to recover sequences related to known genes and does not select for complete gene sequences and functional products [9]. Function-driven analysis depends on the detection of a specific phenotype expressed by a clone. To be successful, it requires transcription of the cloned genes and efficient translation by the heterologous screening host, which are the main limitations of the function-driven approach [18]. Expression can be driven by the vector’s host-specific promoter in plasmid libraries, but limited in cosmid, fosmid or BAC libraries due to the size of the DNA insert. Nevertheless, those large-insert libraries cover longer DNA sequences with fewer clones and are more appropriate to recover complex pathways encoded by large gene clusters.
The detection of a target gene in a metagenomic library often involves screening many thousands of clones. To increase the chances of finding positive clones, it is possible to generate a laboratory enrichment culture from an environmental sample, prior to DNA isolation. By using particular growth conditions, it is possible to increase the proportion of organisms harboring the target trait [13–15]. However, this must be balanced against the overall loss of population diversity [11].
The Escherichia coli T7 expression system developed by Studier and Moffatt [38] is commonly used to achieve high-level protein production. The system is based on the T7 bacteriophage RNA polymerase (T7 RNApol), which directs the selective transcription of genes cloned downstream of the major T7 late promoter. The T7RNApol is characterized by very high activity, elongating messenger RNA (mRNA) chains about five times faster than the E. coli RNA polymerase [17]. The phage RNA polymerase can also generate very long mRNAs and is poorly terminated by unrelated transcription terminators [31]. Since the original publication of Studier and Moffatt, the T7 expression system has been adapted to mammalian cells and several bacteria [3, 6, 10, 12, 16, 21, 22]. Lussier et al. [29] have developed a bifunctional vector, pFX583, that allows T7 RNApol-directed transcription in E. coli and in the filamentous Gram-positive bacterium Streptomyces lividans. As pFX583 features a λ cos sequence, it can be used as a cosmid vector for cloning of large DNA fragments. The different characteristics of pFX583 make it very attractive for the construction of metagenomic libraries and for function-driven screening. To use with this vector, a S. lividans strain that inducibly produces T7 RNApol was also developed by the same group. S. lividans is known as a very useful alternative host to expand the number of genes detected in metagenomic function-based screening [30].
Here, in order to isolate new lipolytic and proteolytic enzymes, a metagenomic cosmid library was constructed with pFX583 using DNA extracted from the biomass of an enriched fed-batch reactor. The resulting library was screened for enzymatic activity in E. coli and S. lividans. The approach described in this paper combined biomass enrichment and multihost screening, strategies known to increase gene detection frequency [9]. The use of pFX583 has allowed T7 RNApol-directed transcription of the cloned metagenomic DNA fragments, which has potentially enhanced the expression of the foreign genes. Many clones with lipolytic activity were detected, from which a new lipase was isolated and partially characterized. For unknown reason, no protease was detected.
Materials and methods
Bacterial strains, culture media, and growth conditions
Bacterial strains and vectors used in this study are listed in Table 1. E. coli strains were grown in 2xTY medium (16 g/l tryptone, 10 g/l yeast extract, 5 g/l NaCl) with or without agar at 37°C. Screening was performed on 2xTY agar supplemented with 1% tributyrin (Sigma) or 2.5% skim milk (Difco). S. lividans was grown at 34°C, and R5 medium [23] supplemented with 2.5% skim milk was used for protoplast regeneration and for direct screening of proteolytic activity. Antibiotics were added to the growth media at the following concentrations: kanamycin at 50 μg/ml or 200 μg/ml, thiostrepton at 50 μg/ml (solid medium) or 5 μg/ml (liquid medium), and apramycin at 50 μg/ml. The culture medium used for biomass enrichment was an adaptation of Basal Salts Medium [33] supplemented with ground meat extract.
Sequencing fed-batch reactor enrichment
A 10-l sequencing fed-batch reactor (SFBR) was inoculated with 10 g black soil, 10 g shrimp compost, 50 ml biomass from a SFBR for phosphor elimination in swine waste, 50 ml biomass from an aerobic thermophilic sequencing batch reactor for swine waste treatment [20], and two swabs from the cafeteria of the INRS-Institut Armand-Frappier. The biomass of the SFBR was enriched with ground meat extract solution as a source of carbon. The solution was prepared with extralean ground beef composed of 10% lipids, 20% proteins, and 0% carbohydrates, based on the product label. In 1 l ion-free water, 100 g meat was homogenized, and pH was adjusted to 7.0 with NaOH. The resulting solution was filtered by 850-μm sieve (20 mesh). The SFBR biomass was submitted to 30 cycles of 72 h in which the pH gradually shifted from 7 to 8.5 and back to 7. Simultaneously, the temperature increased from 50°C to 70°C and returned to 50°C.
Library construction
The DNA used for the construction of the metagenomic library was obtained from the biomass of the SFBR. The method used to extract high-molecular-weight DNA included lysozyme digestion, freeze/thaw cycles, and phenol/chloroform extractions. Total DNA was partially digested with BamHI to generate fragments of 35–45 kb, and ligated with pFX583 linearized with the same restriction enzyme. The concatemeric DNA was packaged using the MaxPlax™ Lambda Packaging Extracts (Epicentre Biotechnologies) and transduced into E. coli RosettaBlue(DE3)/pLysS (Novagen).
Functional-based screening and gene isolation
Transductants were replica-plated on 2xTY agar supplemented with tributyrin or skim milk and 0.4 mM isopropyl β-d-1-thiogalactopyranoside (IPTG). After 48 h of incubation at 37°C, colonies showing a zone of clearance indicated lipolytic or esterolytic activities. The cosmids from the positive colonies were extracted for analysis. To isolate the putative lipase or esterase encoding genes, a subcloned library was generated in the plasmid pCR4-TOPO using the TOPO Shotgun subcloning kit (Invitrogen). The subcloned library was rescreened in E. coli TOP10 (Invitrogen), and the plasmids from positive colonies were sequenced at the McGill University and Génome Québec Innovation Centre.
Partial enzymatic characterization
Enzymatic assays were done with supernatant from an overnight-grown culture of the E. coli TOP10 clone containing lipF5–11, which was obtained from the subcloned library. A clone with an empty plasmid was used as negative control. Assays were performed at 50°C in a 96-well microplate with 5 μl of supernatant in a final volume of 100 μl. The reaction mix contained 1 mM of a particular p-nitrophenyl (pNP) acyl ester (Sigma) in 50 mM Tris–HCl pH 8, 0.05% (m/v) CaCl2, 0.5% (v/v) Triton X-100. Absorbance was read at 405 nm after 48 min of incubation with a Fusion microplate reader (Packard). Substrates with different acyl chain length were tested: pNP-acetate (C2), pNP-butyrate (C4), pNP-caproate (C6), pNP-caprylate (C8), pNP-caprate (C10), pNP-laurate (C12), pNP-myristate (C14), pNP-palmitate (C16), and p-nitrophenyl stearate (C18). The influence of pH was tested with pNP-palmitate using 50 mM Tris–HCl buffer for pH 6, 7, and 8 and with 50 mM glycine-NaOH buffer for pH 9, 10, and 11. The thermostability of LipF5–11 was evaluated by pre-incubating the culture supernatant for 2 h at temperatures ranging between 50°C and 70°C. The enzymatic assay was then conducted as described above.
Results
Metagenomic library construction
The library was constructed using genomic DNA extracted from the biomass of a sequencing fed-batch reactor enriched for bacteria able to grow at alkaline pH and temperatures ranging between 50°C and 70°C. The genomic DNA was cloned in the vector pFX583, which featured a λ cos sequence, a T7 promoter, and a kanamycin/neomycin selection marker. Because of the pMB1 and pJV1 replicons of pFX583, the library can be screened in E. coli and Streptomyces species.
Function-based screening
The packaged library was transduced into E. coli RosettaBlue(DE3)/pLysS, a recA strain harboring the T7RNApol encoding gene under the control of the lacUV5 promoter. Transductants were replica-plated on 2xTY agar supplemented with tributyrin and skim milk. From approximately 2,000 screened colonies, 17 showed esterolytic/lipolytic activity, but no proteolytic activity was detected. The same number of cosmids was transferred to Streptomyces lividans 10T7 by protoplast transformation. Approximately 10,000 colonies were screened on R5 agar supplemented with skim milk. As for E. coli, no colonies showed proteolytic activity.
Subcloning and sequence analysis
Cosmids from positive clones were sheared by nebulization [39] into 1–6-kb fragments and subcloned into pCR4-TOPO via topoisomerase-directed ligation. The resulting libraries were introduced in E. coli TOP10, and plasmids from clones showing activity were sequenced. Nucleotide sequences were analyzed by BLASTX (NCBI) using the nonredundant protein sequences database. Some of the clones contained lipase genes previously isolated from another metagenomic library constructed by our group [32]. One of the plasmids contained a new secreted lipase-encoding gene (lipF5–11) (GenBank accession no. HQ009871) showing the highest similarity with predicted lipases from Streptomyces pristinaespiralis (EDY64232), Thermomonospora curvata (ACY99099), and Conexibacter woesei (ADB52323), with identities of 42%, 39%, and 39%, respectively. Downstream of lipF5–11, the plasmid also contained two open reading frames (ORFs) that showed homology with PpiC-type peptidyl-prolyl cis–trans isomerase and lipase chaperone genes. The amino acid sequence of LipF5–11 featured a predicted signal peptide (residues 1–26) and the conserved pentapeptide Ala-Xaa-Ser-Xaa-Gly (residues 99–103) typical of lipases from Bacillus [2] (Fig. 1). Multiple sequence alignment performed with ClustalW2 [25] also revealed a putative oxyanion hole sequence (H34-G35) and the catalytic residues Asp171 and His193.

Multiple amino acid sequence alignment performed with ClustalW2. LipF5–11 was aligned with the three putative lipases that showed the highest similarity and with a lipase from B. subtilis. Streptomyces pristinaespiralis (EDY64232), Thermomonospora curvata (ACY99099), Conexibacter woesei (ADB52323), and Bacillus subtilis (AAA22574.1). Lower case indicates the predicted signal peptide; the putative oxyanion hole (HG), the pentapeptide (AHSNG) as well as the aspartic acid and the histidine as putative residues of the active site are underlined. “*” means that the residues in that column are identical in all sequences in the alignment. “:” means that conserved substitutions have been observed. “.” means that semiconserved substitutions are observed
Partial biochemical characterization
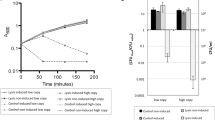
A high-throughput assay was used for fast partial characterization of the unpurified lipolytic enzyme LipF5–11. The results presented here are representative of what was obtained in independent assays, although the global lipolytic activity of the supernatant could vary. To evaluate the substrate specificity of the enzyme, pNP-acyl esters with carbon chain length varying between 2 and 18 were tested (Fig. 2). Hydrolysis was detected for all substrate tested, but was clearly more important for the long-chain pNP-acyl esters. Optimal pH was determined by incubating crude protein extract with pNP-palmitate (C16) at pH ranging from 6 to 11. LipF5–11 was most active between pH 6 and 9, with highest activity at pH 8 (Fig. 3). Thermostability was evaluated by looking at the residual lipolytic activity in the supernatant after pre-incubation of 2 h at temperatures ranging from 50°C to 70°C (Fig. 4). Pre-incubation below 55°C did not have an important effect on the activity of LipF5–11. More than 50% of the activity remained after 2 h at 60°C, but it dropped to 5% at 70°C.

Effect of chain length on the activity of LipF5–11. Assays were conducted at 50°C in 50 mM Tris–HCl pH 8.0. Absorbance measured at 60 min was used for comparison. pNP-acetate (C2), pNP-butyrate (C4), pNP-caproate (C6), pNP-caprylate (C8), pNP-caprate (C10), pNP-laurate (C12), pNP-myristate (C14), pNP-palmitate (C16), and p-nitrophenyl stearate (C18)

Effect of pH on the activity of LipF5–11. Lipase activity was assayed toward pNP-palmitate (C16) at 50°C in 50 mM Tris–HCl buffer for pH 6, 7, and 8 and in 50 mM glycine-NaOH buffer for pH 9, 10, and 11

Thermostability assay. Activity of LipF5–11 was assayed after pre-incubation of 2 h at temperatures ranging from 50°C to 70°C
Discussion
In the search for novel biocatalysts or molecules, function-driven screening is the only strategy that has the potential to identify completely new genes [9]. However, the functional approach is limited by its reliance on adequate expression of the cloned genes in a surrogate host. Aside from the codon usage of the foreign genes, the regulation of the promoters and their recognition by the host RNA polymerase can also impair the detection of positive clones. In the present study, a metagenomic library was constructed with the bifunctional cosmid vector pFX583. The T7 promoter/terminator in pFX583 allows the use of the highly effective T7 RNApol to transcribe the cloned DNA. Combined with a T7 RNApol-producing host, pFX583 has the potential to increase the number of colonies with positive activity. A similar approach was successfully used by Leggewie et al. [26], using a transposon harboring bidirectional T7 promoters. Introducing this transposon in a cosmid library allowed inducible expression of its flanking regions in both directions, thus enhancing detection of clones with lipolytic activity. However, this transposon approach normally requires extraction of the cosmid library, in vitro transposition, followed by re-introduction into the screening host. The diversity of the library with transposon is therefore dependent on the number of cosmids extracted at first. Also, because the library with transposon is reintroduced into the screening host, repetition can occur and will require the screening of an extra number of clones to be sure of covering the starting clones.
The number of screened clones required to recover genes of interest is directly linked to the size of the cloned DNA, but also to the frequency of organisms with the desired activity in the biomass from which DNA is extracted. To increase this frequency, biomass can be subjected to conditions favoring microorganisms harboring the desired traits, such as carbon or nitrogen sources, pH, and temperature. However, the enrichment step can reduce the genetic diversity by promoting fast-growing and culturable members of a microbial consortia [9]. If conducted too extensively, it can even decrease the probability of discovering new genes [11]. Nevertheless, well-monitored enrichment combined with diversified starting material is an efficient strategy to increase the number of positive clones in a metagenomic library screen [8, 15, 24, 32, 34]. Also, the cultivation step generally facilitates isolation of high-quality DNA. In the current study, the DNA used for the construction of the metagenomic library was extracted from a biomass enriched for microorganisms capable of growing at high temperature and pH between 7 and 8.5. Also, inoculums were from various sources, ensuring good starting diversity.
The metagenomic library was first introduced in the common host E. coli. The strain used, E. coli RosettaBlue(DE3), is a recA1 T7 RNApol-producing strain. With this strategy, approximately 2,000 clones were screened for esterolytic/lipolytic and proteolytic activity. From a small number of clones, compared with most metagenomic screening studies [28], 17 colonies showed cleared zone on tributyrin agar. The high success rate obtained in this functional screening experiment showed that the enrichment step was properly conducted. The use of ground beef extract was likely a good source of carbon for the selection of bacteria producing lipolytic enzymes. Also, the cosmid pFX583 used for the construction of the metagenomic library and the E. coli RosettaBlue(DE3)/pLysS strain allowed efficient detection of esterase/lipase enzymes, whose expression could be driven by foreign promoters and the T7 promoter of the vector. Despite the great success of the lipolytic enzymes screen, no proteolytic activities were detected for the same number of clones.
Although E. coli is the most employed host for functional screening [8], using an alternative host proved to be useful to expand the range of detected genes [7, 27, 30, 42, 43]. Bacteria with different codon usage and higher protein secretion capacity than E. coli are of particular interest, as these characteristics can greatly influence the result of function-based screens. Because of the bifunctionality of the pFX583 vector, it was possible to screen the library in S. lividans 10T7. Like E. coli RosettaBlue(DE3), S. lividans 10T7 allows T7RNApol-directed transcription [29]. This Gram-positive host also has very high secretion capacity and can produce the foreign protein directly into the culture medium [4]. Unfortunately, screening in S. lividans 10T7 gave results similar to E. coli and failed to detect proteolytic activity. This disappointing result contrasts with the high number of positive hits obtained in the lipolytic activity screen. Although proteolytic enzymes are present in all bacteria, intracellular proteases are highly specific [5] and may not be detected on skimmed milk agar, even if the cells are lysed. On the other hand, extracellular proteases are mostly produced as inactive precursors that require to be activated by limited proteolysis [41]. The activation step could be suboptimal in a heterologous host and therefore affect the detection of the enzyme. Also, the skimmed milk agar method may not be sensitive enough for detection of small amount of proteases. Still, only a small number of clones were screened compared with most metagenomic studies [37], and a more extensive screen could possibly allow the isolation of a protease encoding gene.
From the lipolytic activity screen in E. coli, a new lipase encoding gene was isolated. LipF5–11 showed only low similarity with available amino acid sequences, the highest identity being 42% for a predicted lipase from Streptomyces pristinaespiralis. The amino acid sequence of LipF5–11 has all the characteristics of a true lipase, featuring the conserved pentapeptide Ala-Xaa-Ser-Xaa-Gly typical of lipases from Bacillus [2]. A high-throughput microplate enzymatic assay was conducted on culture supernatant to evaluate substrate specificity, optimal pH, and thermostability. LipF5–11 had highest activity for pNP-acyl esters with long carbon chains (>10), indicating that it is probably a lipase and not an esterase [19, 40]. However, a natural substrate should be used to confirm the true substrate specificity of LipF5–11 [36]. In accordance with the conditions used for biomass enrichment, LipF5–11 showed highest activity at pH 8. It also retained more than 50% of its activity after pre-incubation of 2 h up to 60°C. Downstream of lipF5–11, sequence analysis of the cosmid revealed putative PpiC-type peptidyl-prolyl cis–trans isomerase and lipase chaperone genes arranged in an operon-type structure. Although the expression of lipF5–11 was sufficient to allow detection and partial characterization, the amount produced was still very low (result not shown). This could be due to improper folding or secretion in E. coli, possibly because the isomerase and the lipase chaperone were not well expressed or nonfunctional. For future purification and characterization, the productivity problem of LipF5–11 will have to be addressed.
In this study, biomass enrichment combined with the use of the vector pFX583 allowed a high yield of lipolytic clones. For the first time, a bifunctional cosmid vector allowing T7RNApol-directed transcription has been used for the construction and screening of a metagenomic library. The cosmid pFX583 has the potential to increase the transcription of the foreign genes and can be used in E. coli and S. lividans. These two characteristics can greatly enhance the gene detection frequency in metagenomic library screening. The approach used in this study led to the identification of a new lipase, a type of biocatalyst with great industrial potential.
References
Amann RI, Ludwig W, Schleifer KH (1995) Phylogenetic identification and in situ detection of individual microbial cells without cultivation. Microbiol Rev 59:143–169
Arpigny JL, Jaeger KE (1999) Bacterial lipolytic enzymes: classification and properties. Biochem J 343:177–183
Barnard GC, Henderson GE, Srinivasan S, Gerngross TU (2004) High level recombinant protein expression in Ralstonia eutropha using T7 RNA polymerase based amplification. Protein Expr Purif 38:264–271
Binnie C, Cossar JD, Stewart DI (1997) Heterologous biopharmaceutical protein expression in Streptomyces. Trends Biotechnol 15:315–320
Bond JS, Butler PE (1987) Intracellular proteases. Annu Rev Biochem 56:333–364
Conrad B, Savchenko RS, Breves R, Hofemeister J (1996) A T7 promoter-specific, inducible protein expression system for Bacillus subtilis. Mol Gen Genet 250:230–236
Courtois S, Cappellano CM, Ball M, Francou FX, Normand P, Helynck G, Martinez A, Kolvek SJ, Hopke J, Osburne MS, August PR, Nalin R, Guerineau M, Jeannin P, Simonet P, Pernodet JL (2003) Recombinant environmental libraries provide access to microbial diversity for drug discovery from natural products. Appl Environ Microbiol 69:49–55
Daniel R (2004) The soil metagenome—a rich resource for the discovery of novel natural products. Curr Opin Biotechnol 15:199–204
Daniel R (2005) The metagenomics of soil. Nat Rev Microbiol 3:470–478
Drepper T, Arvani S, Rosenau F, Wilhelm S, Jaeger KE (2005) High-level transcription of large gene regions: a novel T(7) RNA-polymerase-based system for expression of functional hydrogenases in the phototrophic bacterium Rhodobacter capsulatus. Biochem Soc Trans 33:56–58
Elend C, Schmeisser C, Leggewie C, Babiak P, Carballeira JD, Steele HL, Reymond JL, Jaeger KE, Streit WR (2006) Isolation and biochemical characterization of two novel metagenome-derived esterases. Appl Environ Microbiol 72:3637–3645
Elroy-Stein O, Moss B (1990) Cytoplasmic expression system based on constitutive synthesis of bacteriophage T7 RNA polymerase in mammalian cells. Proc Natl Acad Sci USA 87:6743–6747
Entcheva P, Liebl W, Johann A, Hartsch T, Streit WR (2001) Direct cloning from enrichment cultures, a reliable strategy for isolation of complete operons and genes from microbial consortia. Appl Environ Microbiol 67:89–99
Ferrer M, Golyshina OV, Chernikova TN, Khachane AN, Martins Dos Santos VA, Yakimov MM, Timmis KN, Golyshin PN (2005) Microbial enzymes mined from the Urania deep-sea hypersaline anoxic basin. Chem Biol 12:895–904
Gabor EM, de Vries EJ, Janssen DB (2004) Construction, characterization, and use of small-insert gene banks of DNA isolated from soil and enrichment cultures for the recovery of novel amidases. Environ Microbiol 6:948–958
Gamer M, Frode D, Biedendieck R, Stammen S, Jahn D (2009) A T7 RNA polymerase-dependent gene expression system for Bacillus megaterium. Appl Microbiol Biotechnol 82:1195–1203
Golomb M, Chamberlin M (1974) Characterization of T7-specific ribonucleic acid polymerase. IV. Resolution of the major in vitro transcripts by gel electrophoresis. J Biol Chem 249:2858–2863
Handelsman J (2004) Metagenomics: application of genomics to uncultured microorganisms. Microbiol Mol Biol Rev 68:669–685
Jaeger KE, Dijkstra BW, Reetz MT (1999) Bacterial biocatalysts: molecular biology, three-dimensional structures, and biotechnological applications of lipases. Annu Rev Microbiol 53:315–351
Juteau P, Tremblay D, Ould-Moulaye CB, Bisaillon JG, Beaudet R (2004) Swine waste treatment by self-heating aerobic thermophilic bioreactors. Water Res 38:539–546
Kang Y, Son MS, Hoang TT (2007) One step engineering of T7-expression strains for protein production: increasing the host-range of the T7-expression system. Protein Expr Purif 55:325–333
Katzke N, Arvani S, Bergmann R, Circolone F, Markert A, Svensson V, Jaeger KE, Heck A, Drepper T (2010) A novel T7 RNA polymerase dependent expression system for high-level protein production in the phototrophic bacterium Rhodobacter capsulatus. Protein Expr Purif 69:137–146
Kieser T, Bibb MJ, Buttner MJ, Chater KF, Hopwood DA (2000) Practical Streptomyces genetics. The John Innes Foundation, Norwich
Knietsch A, Waschkowitz T, Bowien S, Henne A, Daniel R (2003) Construction and screening of metagenomic libraries derived from enrichment cultures: generation of a gene bank for genes conferring alcohol oxidoreductase activity on Escherichia coli. Appl Environ Microbiol 69:1408–1416
Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG (2007) Clustal W and Clustal X version 2.0. Bioinformatics 23:2947–2948
Leggewie C, Henning H, Schmeisser C, Streit WR, Jaeger KE (2006) A novel transposon for functional expression of DNA libraries. J Biotechnol 123:281–287
Li Y, Wexler M, Richardson DJ, Bond PL, Johnston AW (2005) Screening a wide host-range, waste-water metagenomic library in tryptophan auxotrophs of Rhizobium leguminosarum and of Escherichia coli reveals different classes of cloned trp genes. Environ Microbiol 7:1927–1936
Lorenz P, Eck J (2005) Metagenomics and industrial applications. Nat Rev Microbiol 3:510–516
Lussier FX, Denis F, Shareck F (2010) Adaptation of the highly productive T7 expression system to Streptomyces lividans. Appl Environ Microbiol 76:967–970
Martinez A, Kolvek SJ, Yip CL, Hopke J, Brown KA, MacNeil IA, Osburne MS (2004) Genetically modified bacterial strains and novel bacterial artificial chromosome shuttle vectors for constructing environmental libraries and detecting heterologous natural products in multiple expression hosts. Appl Environ Microbiol 70:2452–2463
McAllister WT, Morris C, Rosenberg AH, Studier FW (1981) Utilization of bacteriophage T7 late promoters in recombinant plasmids during infection. J Mol Biol 153:527–544
Meilleur C, Hupe JF, Juteau P, Shareck F (2009) Isolation and characterization of a new alkali-thermostable lipase cloned from a metagenomic library. J Ind Microbiol Biotechnol 36:853–861
Reddy CA, Beveridge TJ, Breznak JA, Marzluf G, Schmidt TM, Snyder LR (2007) Methods for general and molecular microbiology, 3rd edn. ASM Press, Washington, DC
Rees HC, Grant S, Jones B, Grant WD, Heaphy S (2003) Detecting cellulase and esterase enzyme activities encoded by novel genes present in environmental DNA libraries. Extremophiles 7:415–421
Schloss PD, Handelsman J (2003) Biotechnological prospects from metagenomics. Curr Opin Biotechnol 14:303–310
Sigurgísladóttir S, Konráòsdóttir M, Jónsson Á, Kristjánsson JK, Matthiasson E (1993) Lipase activity of thermophilic bacteria from icelandic hot springs. Biotechnol Lett 15:361–366
Steele HL, Jaeger KE, Daniel R, Streit WR (2009) Advances in recovery of novel biocatalysts from metagenomes. J Mol Microbiol Biotechnol 16:25–37
Studier FW, Moffatt BA (1986) Use of bacteriophage T7 RNA polymerase to direct selective high-level expression of cloned genes. J Mol Biol 189:113–130
Surzycki S (2000) Basic techniques in molecular biology. Springer, Berlin
Verger R (1997) ‘Interfacial activation’ of lipases: facts and artifacts. Trends Biotechnol 15:32–38
Wandersman C (1989) Secretion, processing and activation of bacterial extracellular proteases. Mol Microbiol 3:1825–1831
Wang GY, Graziani E, Waters B, Pan W, Li X, McDermott J, Meurer G, Saxena G, Andersen RJ, Davies J (2000) Novel natural products from soil DNA libraries in a streptomycete host. Org Lett 2:2401–2404
Wexler M, Bond PL, Richardson DJ, Johnston AW (2005) A wide host-range metagenomic library from a waste water treatment plant yields a novel alcohol/aldehyde dehydrogenase. Environ Microbiol 7:1917–1926
Acknowledgments
This work was supported by a Strategic Grant from the Natural Sciences and Engineering Council of Canada. The authors thank Raymonde Jetté and July Dorion-Thibaudeau for their technical assistance.
Author information
Authors and Affiliations
Corresponding author
Additional information
The authors François-Xavier Lussier and Olivier Chambenoit contributed equally to this work.
Rights and permissions
About this article
Cite this article
Lussier, FX., Chambenoit, O., Côté, A. et al. Construction and functional screening of a metagenomic library using a T7 RNA polymerase-based expression cosmid vector. J Ind Microbiol Biotechnol 38, 1321–1328 (2011). https://doi.org/10.1007/s10295-010-0915-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10295-010-0915-2