
Abstract
A unique cationic polyglucosamine biopolymer PGB-1 comprising more than 95% D-glucosamine was excretively produced from a new bacterial strain Enterobacter sp. BL-2 under acetate-mediated culture conditions. Since the biopolymer PGB-1 could be synthesized from the UDP-N-acetylglucosamine monomer derived from the hexosamine pathway, three glmS, glmM, and glmU genes in the hexosamine pathway were cloned from Enterobacter sp. BL-2, and their molecular structures were elucidated. The cloned glmS, glmM, and glmU genes were reintroduced into the parent strain Enterobacter sp. BL-2 through a conjugative transformation for the overproduction of the biopolymer PGB-1. The biopolymer production increased 1.5-fold in the transconjugant Enterobacter sp. BL-2S over-expressing the first-step glmS gene encoding glucosamine-6-phosphate synthase. The transconjugant Enterobacter sp. BL-2S was cultivated pH-stat fed-batch widely, while intermittently feeding an acetate solution to maintain a constant pH level of 8.0 for 72 h, resulting in 1.15 g/L of the extracellular polyglucosamine biopolymer PGB-1.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Chitin [ß(1→4) linked poly-N-acetylglucosamine] and chitosan [ß(1→4) linked poly-D-glucosamine] are the major cell-wall components of yeast and fungi, and also represent the typical microbial polyglucosamines polymerized from the UDP-N-acetylglucosamine (UDP-GlcNAc) monomer derived from the hexosamine pathway [8, 17]. Recently, several polyglucosamine biopolymer excretively produced by bacterial strains under acetate-mediated culture conditions have been investigated, including the bioflocculant from Citrobacter sp. TKF04 [6, 10], microbial polyglucosamine biopolymers PGB-1 from Enterobacter sp. BL-2 [20], and PGB-2 from Citrobacter sp. BL-4 [11].
In particular, the microbial polyglucosamine biopolymers PGB-1 and 2, previously investigated by current authors [11, 20] were found to contain more than 95% D-glucosamine, and show similar FT-IR and NMR spectra to chitosan from crab shells. Therefore, these unique extracellular microbial polyglucosamine biopolymers may be potential substitutes for chitin and chitosan that are widely used in the food, pharmaceutical, and agricultural industries.
Bacteria are also known to synthesize the cell-wall components peptidoglycan and lipopolysaccharide using UDP-GlcNAc derived from the well-known hexosamine biosynthesis pathway [1, 14]. Three enzymes: (1) glucosamine-6-phosphate (GlcN-6-P) synthase encoded by glmS [4], (2) phosphoglucosamine mutase encoded by glmM [16], and (3) the unique bifunctional GlcN-1-P acetyltransferase and GlcNAc-1-P uridyltransferase encoded by glmU [15] have already been documented in Escherichia coli. In addition, the extracellular polyglucosamine biopolymer PGB-1 would also seem to be synthesized via the same hexosamine pathway in Enterboacter sp. BL-2, but only actively under acetate-mediated culture condition.
In this work, three glmS, glmM, and glmU genes were newly cloned from Enterobacter sp. BL-2 [20], and their molecular structures were elucidated. The cloned genes were reintroduced in the parent Enterobacter sp. BL-2 using a conjugative transformation to control the metabolic flux of the intermediates derived from the hexosamine pathway to the biosynthesis of the biopolymer PGB-1. The transconjugant Enterobacter sp. BL-2S over-expressing the glmS gene encoding GlcN-6-P synthase was cultivated pH-stat fed-batch wisely to achieve the overproduction of the extracellular polyglucosamine biopolymer PGB-1.
Materials and methods
Bacterial strains
The parent strain Enterobacter sp. BL-2 was reported in our previous work [20]. The strain was cultivated in a basal liquid medium composed of 0.1% (w/v) yeast extract, 0.1% (NH4)2SO4, 2.0% MgSO4·7H2O, 0.5% NaCl, and 0.05% trace elements [6, 20], supplementing 1.5% sodium acetate as the carbon source at 30 °C, pH 7.5 for 48 h.
Cloning of glmS, glmM, and glmU genes in Enterobacter sp. BL-2
The glmS, glmM, and glmU genes in the genomic DNA isolated from Enterobacter sp. BL-2 were amplified by a PCR using the primer sets designed from genomic databases of E. coli K-12 (http://www.ecocyc.org); S-f (5′-GGCGACGATGTGTTTGTTGG-3′) and S-r (5′-TTACTCAACCGTAACCGATTTTGC-3′) for glmS, M-f (5′-ATTGTTGCTCGACCCCGG-3′) and M-r (5′-TTAAACGGCTTTTACTGCATCGGCG-3′) for glmM, and U-f (5′-TGTTCTCAAATTA CAGTCAGGACGC-3′) and U-r (5′-GATCGCGCCAACAATTCCACA-3′) for glmU. The glmS, glmM, and glmU genes were cloned into a cloning pGEM®-T Easy vector (Promega, Madison, WI, USA), sequenced using an Automatic Sequencer ABI 377 (Applied Biosystems, Foster City, CA, USA), and the nucleotide sequences analyzed using the BLAST program at NCBI.
Construction of conjugative plasmid and transformation of glmS, glmM, and glmU genes in Enterobacter sp. BL-2
The glmS, glmM, and glmU genes were transformed into Enterobacter sp. BL-2 using a conjugation method. The conjugation vector pBBR1MCS-2T was constructed by inserting the oriT (conjugation-mediated plasmid transfer) gene separated from pEX100T [19] to be recognized by the RP4 transfer gene in the donor E. coli S17-1, into the BmgBI blunt digestion site of pBBR1MCS-2 [12], as depicted in Fig. 1. The glmS and glmM gene fragments in the pGEM®-T Easy vector were then double-digested by SacI and ApaI, while the glmU gene was digested by EcoRI before inserting into the conjugation vector pBBR1MCS-2T to obtain the conjugative plasmids pBBR1MCS-2TS, 2TM, and 2TU, respectively.

Construction of conjugative vector pBBR1MCS-2T and plasmids pBBR1MCS-2TS, 2TM, and 2TU carrying glmS, glmM, and glmU, respectively
The conjugative plasmids were then transformed into the donor E. coli S17-1 by heat-shock, followed by conjugative transformation, where the donor E. coli S17-1 cells, numbering 5 × 107, and the recipient Enterobacter sp. BL-2 cells, numbering 2 × 108, were dropped onto nitrocellulose filters on an LB plate, and mated at 32 °C for 12 h. Thereafter, the transconjugants were suspended in a fresh LB liquid medium, and spread on an LB plate containing 50 μg/mL kanamycin as the selection marker. Finally, three transconjugants Enterobacter sp. BL-2S, 2M, and 2U amplifying glmS, glmM, and glmU, respectively, were selected after cultivation at 28 °C for 10 h.
Real-time PCR of glmS, glmM, and glmU genes
The cDNA was synthesized by the TaqMan reverse transcription reagent (Applied Biosystems) using the total RNA isolated by an RNA STAT-60™ solution (Tel-Test Inc., Friendswood, TX, USA) as the template. The primer sets were designed using the sequences of the glmS, glmM, and glmU genes as; SRT-f : 5′-CCAAACGCGATCAAAAACAC-3′; SRT-r : 5′-TCAACCTTGCCGAGCAGTT-3′; MRT-f : 5′-ATATGAGCCTGCACGACCTTTG-3′; MRT-r : 5′-TGACTCATGCTCAAGTGGATCG-3′; URT-f : 5′-CGTGGAAATGAAAAAAGCGC-3′, URT-r : 5′-TCCTGCACCAATATTCACGTTG-3′, respectively.
The transcription level was analyzed using a 2×SYBR Green Master Mix (Applied Biosystems, Foster City, CA, USA). The thermal profile for the SYBR RT-PCR was 1 cycle at 50 °C for 2 min to activate the SYBR green dye and 95 °C for 10 min for denaturation of the cDNA, followed by 40 cycles at 95 °C for 15 s and 60 °C for 15 s to amplify the target cDNA using the 16S rRNA gene, previously reported [20], as the standard transcription marker.
Measurement of fructose-1,6-bisphosphatase, isocitrate lyase, and glcN-6-P synthase activities
The cell was washed and re-suspended in a cold 0.1 M Hepes buffer (pH 7.1) containing 100 mM KCl, 1 mM EDTA, 1 mM dithiothreitol, and 1 mM phenylmethyl-sulfonylfluoride before sonication (30 × 5 s pulses with 5 s intervals) using a Ultraschallprozessor UP 400s (Hielscher, Germany). The fructose-1,6-bisphosphatase (FBPase) activity was determined by measuring the NADPH generated by coupling from the F-6-P formation of FBPase to reduce the NADP in the presence of PGI and G6PDH [5]. A cell extract of 0.1 mL was added to 0.4 mL of an assay mixture containing 20 mM Hepes (pH 7.1), 75 mM KCl, 1 mM EDTA, 10 mM MgSO4, 5 mM F-1,6-BP, and 1 μg PGI/G6PDH. The reaction was initiated at 37 °C for 10 min, and denatured by heating at 90 °C for 5 min, and then the amount of NADPH was measured at 340 nm.
The isocitrate lyase (ICL) activity was determined using the method of Giachetti et al. [7] based on measuring the osazone derivatives produced from phenylhydrazone and glyoxylate generated during the cleavage of isocitrate by ICL. A cell extract of 0.1 mL was added to 0.4 mL of an ICL activity assay mixture composed of 20 mM Hepes, (pH 7.1), 200 mM KCl, 2 mM EDTA, 10 mM MgSO4, 200 mM phenylhydrazone, and 100 mM isocitrate, and then the osazone derivatives was measured at 324 nm.
The GlcN-6-P synthase activity was measured by adding 0.1 mL of the cell extract into 0.4 mL of an assay mixture containing 20 mM Hepes (pH 7.1), 75 mM KCl, 1 mM EDTA, 10 mM F-6-P, and 10 mM glutamine. The reaction was carried out at 37 °C for 30 min, and then stopped by heating at 90 °C for 5 min followed by mixing with 0.5 mL of water. The amount of GlcN-6-P was determined using the modified Elson–Morgan method [9]. The protein was measured using the Bradford method, and 1 U of intrinsic enzyme activity defined as the amount of enzyme catalyzing 1 μm of product per min per mg of cellular protein.
pH-stat fed-batch cultivation of transconjugant Enterobacter sp. BL-2S
The transconjugant Enterobacter sp. BL-2S amplifying the glmS gene was cultivated in a basal liquid medium containing 1.5% sodium acetate at 30 °C and pH 7.5 in a 2.5-L jar-fermenter batch-wisely until the medium pH changed to 8.0. The pH-stat fed-batch culture was initiated based on the intermittent feeding of a stock solution containing 3M acetic acid/ammonium acetate (9:1) to maintain a constant pH level of 8.0 for 72 h, while using 50 μg/mL of kanamycin as the selective pressure.
Measurement of polyglucosamine biopolymer PGB-1
The extracellular polyglucosamine biopolymer PGB-1 in the culture broth was determined using the modified Elson–Morgan method [9], with glucosamine as the standard, after purifying and hydrolyzing in 6 M HCl.
Results and discussion
Flow of acetate in Enterobacter sp. BL-2 for biosynthesis of polyglucosamine biopolymer PGB-1
The carbon utilization patterns in Enterobacter sp. BL-2 were compared after cultivation in a basal medium containing acetate or glucose as the sole carbon source. As shown in Fig. 2, the cells grew well in both acetate and glucose media, although the microbial polyglucosamine biopolymer PGB-1 was only excreted in the acetate medium.

Cultivation of Enterobacter sp. BL-2 in basal medium supplemented with acetate (a) and glucose (b). Biomass concentration (open circle) and extracellular polyglucosamine PGB-1 concentration (filled diamond)
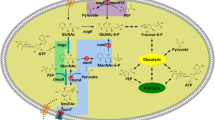
The metabolic flux of acetate as the carbon source in the biosynthesis of the microbial polyglucosamine biopolymer PGB-1 is depicted in Fig. 3. The acetyl-CoA converted from acetate is further metabolized via two ways: the TCA cycle for NADH and ATP generation, or the glyoxylate cycle that generates oxaloacetate, which eventually flows to phosphoenolpyruvate (PEP) and fructose-1,6-bisphosphate (F-1,6-BP) via the EMP pathway, and then F-1,6-BP irreversibly converts into fructose-6-phosphate (F-6-P) by fructose-1,6-bisphosphatase (FBPase). The F-6-P then flows to either gluconeogenesis for the central metabolism or the hexosamine pathway for the biosynthesis of bacterial cell-wall components or the extracellular polyglucosamine biopolymer PGB-1.

Metabolic flux of acetate for biosynthesis of extracellular polyglucosamine biopolymer PGB-1. ① GlcN-6-P synthase encoded by glmS, ② phosphoglucosamine mutase by glmM, ③ bifunctional GlcN-1-P acetyltransferase and GlcNAc-1-P uridyltransferase by glmU, and

To elucidate the flow of acetate, the intrinsic activities of the key enzymes in the above pathways were measured, including ICL in the glyoxylate cycle, FBPase in the EMP pathway, and GlcN-6-P synthase in the hexosamine pathway. As shown in Table 1, the intrinsic activity of ICL only appeared in the acetate medium, indicating that the glyoxylate cycle was only turned on under the acetate-mediated culture conditions. The intrinsic activity of FBPase converting F-1,6-BP to F-6-P also irreversibly increased around 2.3-fold in the acetate medium.
The intrinsic GlcN-6-P synthase, known as the first-step regulatory enzyme in the hexosamine pathway, was also activated in the acetate medium, but only slightly with a 1.3-fold increment compared to that in the glucose medium. The comparatively lower increment of GlcN-6-P synthase, even under the acetate-mediated culture conditions, indicated that the amplification of the genes in the hexosamine pathway potentially contributed to the metabolic flux of intermediates derived from the hexosamine pathway for the biosynthesis of the polyglucosamine biopolymer PGB-1.
Isolation and identification of glmS, glmM, and glmU genes in hexosamine pathway of Enterobacter sp. BL-2
Three genes in the hexosamine pathway, glmS encoding GlcN-6-P synthase, glmM encoding phosphoglucosamine mutase, and glmU encoding bifunctional GlcN-1-P acetyltransferase and GlcNAc-1-P uridyltransferase, were cloned from Enterobacter sp. BL-2, and then registered in the GenBank (EF123214, 123215, and 123216, respectively) as new nucleotide sequences. The homologies of the deduced amino acid sequences were compared with those of other bacterial strains belonging to the Enterobacteriaceae family as shown in Table 2. The molecular weights of the proteins encoded by glmS, glmM, and glmU were 66.7, 47.5, and 49.2, respectively, and showed the close identity with those from E. coli K-12, Shigella boydii Sb277, and Salmonella typhimurium LT2 among Enterobacteriaceae family.
The catalytic domains of three enzymes were also analyzed using BLAST. The first enzyme GlcN-6-P synthase was composed of one glutamine aminotransferase class-II (GFAT) domain and two sugar isomerase (SIS) domains; the second enzyme, phosphoglucosamine mutase, contained four phosphglucomutase/phosphomannomutase (PGM/PMM) domains; and the third enzyme, bifunctional GlcN-1-P acetyltransferase and GlcNAc-1-P uridyltransferase, contained one nucleotidyl (NTP) transferase domain and two left-handed parallel beta helix (LbetaH) domains, similar to other bacterial strains (data not shown).
Expression of glmS, glmM, and glmU genes in Enterobacter sp. BL-2 by conjugative transformation
The cloned new glmS, glmM, and glmU genes were transformed into the parent strain Enterobacter sp. BL-2 through a conjugative transformation, rather than the commonly used heat-shock or electroporation method for Gram-negative bacteria, to avoid the invalidness in Enterobacter sp. BL-2. The transformation of the glmS, glmM, or glmU gene into the parent strain Enterobacter sp. BL-2 was confirmed by a real-time PCR, as shown in Fig. 4. The three glmS, glmM, and glmU genes were well over-expressed in their respective transconjugants Enterobacter sp. BL-2S, 2M, and 2U, and the conjugative plasmids carrying each gene more than 95% stable in each transconjugant after 5 days (data not shown).

Real-time PCR for over-expression of glmS (a), glmM (b), and glmU (c) in Enterobacter sp. BL-2. The Enterobacter sp. BL-2 and the transconjugants were cultivated in an LB medium at 30 °C for 15 h. Data represent the average value of three independent experiments
Effects of over-expressing glmS, glmM, and glmU genes on excretion of polyglucosamine PGB-1
Three transconjugants Enterobacter sp. BL-2S, 2M, and 2U over-expressing glmS, glmM, and glmU genes, respectively, were cultivated under the acetate-mediated culture conditions for 48 h. Concomitantly, the over-expressed glmS, glmM, and glmU genes slightly stimulated cell growth, as shown in Fig. 5, possibly due to the active biosynthesis of the cell-wall components.

Effect of over-expressed glmS, glmM, and glmU genes on cell growth (a) and excretion of polyglucosamine biopolymer PGB-1 (b). Parent strain (open circle), transconjugant Enterobacter sp. BL-2S (filled diamond), BL-2M (filled square), and BL-2U (filled inverted triangle). The cells were cultivated in a basal medium containing 1.5 % sodium acetate at 30 °C for 48 h. Data represent the average value of three independent experiments
However, the excretion of the polyglucosamine biopolymer PGB-1 was influenced in a quite different manner according to the over-expressed glmS, glmM, and glmU genes. The production of the biopolymer PGB-1 was noticeably increased around 1.5-fold from 0.06 to 0.09 g/L by the first-step glmS gene encoding GlcN-6-P synthase, whereas, it was severely depressed to 0.02 g/L by the second-step glmM gene encoding phosphoglucosamine mutase, possibly due to the deficiency of glucose-1,6-diphosphate known as an allosteric activator for phosphoglucosamine mutase as observed in E. coli [16].
In yeast and fungi, the first-step GlcN-6-P synthase in the hexosamine pathway is known to be regulated by chitin in cell-wall [3, 18]; however, the regulation mechanism of the hexosamine pathway in bacteria has not been fully elucidated. The first-step GlcN-6-P synthase encoded by glmS gene also seemed to play a regulatory role on the biosynthesis of the polyglucosamine biopolymer PGB-1 in Enterobacter sp. BL-2.
pH-Stat fed-batch cultivation of transconjugant Enterobacter sp. BL-2S amplifying glmS for excretive over-production of biopolymer PGB-1
The transconjugant Enterobacter sp. BL-2S over-expressing glmS was cultivated pH-stat fed-batch wisely, while intermittently feeding a stock solution containing 3M acetic acid/ammonium acetate (9:1), to maintain a constant pH level of 8.0. As shown in Fig. 6, the biomass concentration slightly increased from 10.10 to 12.05 g/L, while the excretion of the polyglucosamine biopolymer PGB-1 increased around 1.5-fold from 0.75 to 1.15 g/L after over-expressing the glmS gene. The GlcN-6-P synthase was strongly activated for the first 24 h, during the active biosynthesis of the biopolymer PGB-1 was initiated.

pH-stat fed-batch cultivation of transconjugant Enterobacter sp. BL-2S over-expressing glmS gene. Cell growth (a), excretion of polyglucosamine biopolymer PGB-1 (b), and intrinsic activity of GlcN-6-P synthase (c). Parent strain (open circle) and transconjugant Enterobacter sp. BL-2S (filled diamond). The strains were cultivated pH-stat fed-batch wisely, using a 3M acetic acid/ammonium acetate (9:1) solution to maintain a pH level of 8.0. Data represent the average value of three independent experiments
The amplification of glmS gene in the hexosamine pathway was found to be an effective strategy for the overproduction of the extracellular polyglucosamine biopolymer PGB-1 in Enterobacter sp. BL-2, although the function of an unknown enzyme participating in the polymerization of UDP-GlcNAc needs to be further elucidated.
References
Anderson RG, Douglas LJ, Hussey H, Baddiley J (1973) The control of synthesis of bacterial cell walls. Interaction in the synthesis of nucleotide precursors. Biochem J 136:871–876
Blattner FR, Plunkett GIII, Bloch CA, Perna NT, Burland V, Riley M, Collado-Vides J, Glasner JD, Rode CK, Mayhew GF, Gregor J, Davis NW, Kirkpatrick HA, Goeden MA, Rose DJ, Mau B, Shao Y (1997) The complete genome sequence of Escherichia coli K-12. Science 277:1453–1474
Bulik DA, Olczak M, Lucero HA, Osmond BC, Robbins PW, Specht CA (2003) Chitin synthesis in Saccharomyces cerevisiae in response to supplementation of growth medium with glucosamine and cell wall stress. Eukaryot Cell 2:886–900
Dutka-Malen S, Mazodier P, Badet B (1988) Molecular cloning and overexpression of the glucosamine synthase gene from Escherichia coli. Biochimie 70:287–290
Fraenkel DG, Pontermoli S, Horecker BL (1966) The specific fructose diphosphatase of Escherichia coli: properties and partial purification. Arch Biochem Biophys 114:4–12
Fujita M, Ike M, Tachibana S, Kitada G, Kim SM, Inoue Z (2000) Characterization of a bioflocculant produced by Citrobacter sp. TKF04 from acetic and propionic acids. J Biosci Bioeng 89:40–46
Giachetti E, Pinzauti G, Bonaccorsi R, Vanni P (1988) Isocitrate lyase: characterization of its true substrate and the role of magnesium ion. Eur J Biochem 172:85–91
Gooday GW (1977) Biosynthesis of the fungal cell wall-mechanism and implication. J Gen Microbiol 99:1–11
Jang JH, Hia HC, Ike M, Inoue C, Fujita M, Yoshida T (2005) Acid hydrolysis and quantitative determination of total hexosamines of an exopolysaccharide produced by Citrobacter sp. Biotechnol Lett 27:13–18
Jang JH, Ike M, Kim SM, Fujita M (2001) Production of a novel bioflocculant by fed-batch culture of Citrobacter sp. Biotechnol Lett 23:593–597
Kim LS, Hong SJ, Son MK, Lee YH (2006) Polymeric and compositional properties of novel extracellular microbial polyglucosamine biopolymer from new strain of Citrobacter sp. BL-4. Biotechnol Lett 28:241–245
Kovach ME, Elzer PH, Hill DS, Robertson GT, Farris MA, Roop II RM, Peterson KM (1995) Four new derivatives of the broad-host-range cloning vector pBBR1MCS, carrying different antibiotic-resistance cassettes. Gene 166:175–176
McClelland M, Sanderson KE, Spieth J, Clifton SW, Latreille P, Courtney L, Porwollik S, Ali J, Dante M, Du F, Hou S, Layman D, Leonard S, Nguyen C, Scott K, Holmes A, Grewal N, Mulvaney E, Ryan E, Sun H, Florea L, Miller W, Stoneking T, Nhan M, Waterston R, Wilson RK (2001) Complete genome sequence of Salmonella enterica serovar Typhimurium LT2. Nature 413:852–856
Mengin-Lecreulx D, Flouret B, van Heijenoort J (1982) Cytoplasmic steps of peptidoglycan synthesis in Escherichia coli. J Bacteriol 151:1109–1117
Mengin-Lecreulx D, van Heijenoort J (1994) Copurification of glucosamine-1-phosphate acetyltransferase and N-acetylglucosamine-1-phosphate uridyltransferase activities of Escherichia coli; characterization of the glmU gene product as a bifunctional enzyme catalyzing two subsequent steps in the pathway for UNP-N-acetylglucosamine synthesis. J Bacteriol 176:5788–5795
Mengin-Lecreulx D, van Heijenoort J (1996) Characterization of the essential gene glmM encoding phosphoglucosamine mutase in Escherichia coli. J Biol Chem 271:32–39
Milewski S, Gabriel I, Olchowy J (2006) Enzymes of UDP-GlcNAc biosynthesis in yeast. Yeast 23:1–14
Ram AFJ, Arentshorst M, Damveld RA, vanKuyk PA, Klis FM, van den Hondel CAMJJ (2004) The cell wall stress response in Aspergillus niger involves increased expression of the glutamine: fructose-6-phosphate amidotransferase-encoding gene (gfaA) and increased deposition of chitin in the cell wall. Microbiology 150:3315–3326
Schweizer HP, Hoang TT (1995) An improved system for gene replacement and xylE fusion analysis in Pseudomonas aeruginosa. Gene 158:15–22
Son MK, Shin HD, Huh TL, Jang JH, Lee YH (2005) Novel cationic microbial polyglucosamine biopolymer from new Enterobacter sp. BL-2 and its bioflocculation efficacy. J Microbiol Biotechnol 15:626–632
Yang F, Yang J, Zhang X, Chen L, Jiang Y, Yan Y, Tang X, Wang J, Xiong Z, Dong J, Xue Y, Zhu Y, Xu X, Sun L, Chen S, Nie H, Peng J, Xu J, Wang Y, Yuan Z, Wen Y, Yao Z, Shen Y, Qiang B, Hou Y, Yu J, Jin Q (2005) Genome dynamics and diversity of Shigella species, the etiologic agents of bacillary dysentery. Nucleic Acids Res 33:6445–6458
Acknowledgment
This work was supported by a grant (20050401034639) from the BioGreen 21 Program, the Rural Development Administration, the Republic of Korea.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Son, MK., Hong, SJ. & Lee, YH. Acetate-mediated pH-stat fed-batch cultivation of transconjugant Enterobacter sp. BL-2S over-expressing glmS gene for excretive production of microbial polyglucosamine PGB-1. J Ind Microbiol Biotechnol 34, 799–805 (2007). https://doi.org/10.1007/s10295-007-0258-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10295-007-0258-9